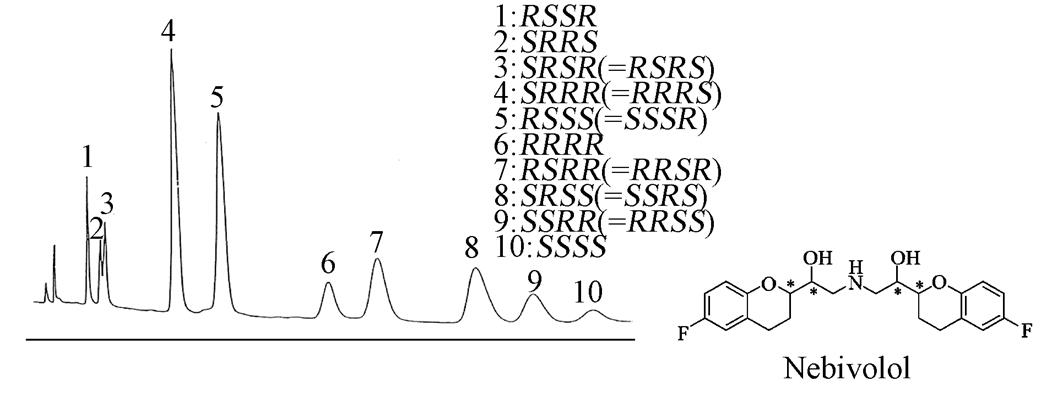
图1 尼比心安HPLC手性分离[16]
Fig. 1 HPLC analysis of nebivolol
吕力琼,步知思,童胜强*
(浙江工业大学药学院,杭州 310014)
摘要:多手性中心药物具有多个立体异构体,空间构型的差异导致异构体之间生物活性差异较大,多手性中心药物的手性分离分析仍然是一个巨大的挑战。关于单手性中心化合物手性分离分析的报道很多,而关于多手性中心药物的手性分离却非常少。本文总结了近年来关于多手性中心化合物色谱手性分离研究进展,主要包括HPLC、GC、毛细管电泳、超临界流体色谱及逆流色谱等在多手性药物分离分析中应用。
关键词:多手性中心;手性分析;高效液相色谱法;毛细管电泳
近年来,手性对映体的分离在医药、农药、食品和化工领域发挥着非常重要的作用,尤其是医药行业,由于安全用药等原因对光学纯活性药物的需求不断增加[1-2]。同一化合物对映体物理化学性质相似,但是药理学和毒理学却完全不同,如(S)-构型多巴为治疗帕金森综合症的首选药物,而(R)-构型则能造成粒状白细胞减少症,使用极度危险;(S)-构型布洛芬是高效非甾体解热镇痛药,而(R)-构型布洛芬基本没有药理活性[3]。含单一手性中心化合物的不同构型之间药效有时相差非常大,而含多手性中心化合物的不同异构体情况更为复杂。如具有双手性中心的拉贝洛尔,拉贝洛尔具有α和β肾上腺素受体阻滞作用,主要用于治疗高血压,临床上用其4种异构体的等比例混合物,其中R,R异构体主要产生非特异性β1和β2阻滞活性,而α1阻滞活性是由S,R异构体产生的,另外2个异构体S,S和R,S也许有活性但很小[4]。盐酸地尔硫卓存在2个手性中心,但是只有(2S,3S)异构体具有药理活性[5]。因此,含多手性中心药物及其中间体的光学异构体手性分离分析等研究工作显得尤为重要。
手性分离方法主要有结晶法、化学拆分、生物拆分和色谱拆分法等,在众多的分离方法中,色谱分离由于其高纯度和高效性得到研究者的青睐。色谱手性拆分原理主要是Dalgliesh在1952年提出的“三点相互作用(three-point interaction)”理论,在1对对映体和手性选择剂之间,为了形成稳定性不同的非对映体分子络合物而达到手性分离的目的,至少需要3个同时发生的分子之间的相互作用力起作用。而且,3个作用力中至少有1个必须是立体化学相互作用,主要包括氢键、偶极-偶极作用、π-π作用、静电作用、疏水作用或空间作用[6]。
多个手性中心化合物的手性分离是手性研究的重要领域。近年来,多手性中心化合物的色谱分析分离主要为HPLC和毛细管电泳等,但是由于分析条件筛选难度大,多数多手性化合物都难以达到理想的分离情况,这类化合物的手性分离仍然是一个巨大的挑战。目前,国外只有少数文献总结关于双手性及多手性中心化合物的手性分离分析[7-10],近年来国内文献尚未有相关研究最新报道。本文主要综述了近年来HPLC、GC、毛细管电泳、超临界流体色谱以及逆流色谱(Countercurrent chromatography,CCC)等方法在多手性中心化合物手性分离中的研究进展。
HPLC手性分离主要有直接法和间接法,直接法可分为手性固定相法(CSP)和手性流动相添加剂法(CMPA),CSP法是由具有光学活性的单体固定在硅胶或其他聚合物上制成手性固定相,通过引入手性环境使对映异构体间呈现物理特征的差异,从而达到光学异构体拆分的目的;CMPA法是在流动相中加入手性添加剂,在普通固定相上吸附,形成短暂的手性环境并对对映异构体间进行手性识别而达到分离,手性试剂主要为环糊精、万古霉素、手性金属配合剂等。间接法为手性试剂衍生法(CDR),对映体混合物在预处理中进行柱前衍生形成1对非对映体,依其物理化学性质的差异,在非手性柱上得以分离[11]。HPLC法具有良好的重现性、选择性、灵敏性、快速性,且其固定相的选择范围广,在手性分离方面发挥着越来越重要的作用[12-14]。虽然HPLC在对映体分离领域发展迅猛,但其在多手性化合物分离方面方法较少,且多为手性固定相法,见表1。1994年,McCarthy[15]首次成功在Chiralpak AD柱上正反相分离萘羟心安。萘羟心安有3个手性中心,有8个异构体,但是在环己烷环上的2个羟基取代基是顺式构型,排除了4个异构体,因而只有4个异构体。在正相条件下,采用正己烷-乙醇-二乙胺(80︰20︰0.4)流动相时,室温下4种对映体都达到了基线分离。尼比心安具有4个手性中心,16个立体异构体(由于存在1个对称面,有些异构体完全相同,存在内消旋形式,因此,仅存在10个立体异构体),1994年Dingenen[16]在Chiralpak AD柱上用乙醇作为流动相,流速0.5 mL·min-1,等度洗脱,检测器波长220 nm,10个异构体能达到基线分离,见图1。茚心安有2个位置异构体,共有4个对映体,Aboul-Enein和Serignese[17]在HPLC上用梯度洗脱的方法在Chiralcel OD柱上分离了茚心安的4种对映体。紧接着,Aboul-Enein[18]团队采用Chiralcel OD柱进一步分离了福莫特罗和拉贝洛尔手性对映体。在流动相为正己烷-二氯乙烷-[乙醇+TFA(20︰1)预混合]-甲醇(58︰35︰7︰0.7),流速0.4 mL·min-1,检测波长308 nm,拉贝洛尔4种对映体得到部分分离,前洗脱的两峰为右旋拉贝洛尔,后两峰为左旋拉贝洛尔;福莫特罗也在条件为正己烷-二氯乙烷-[乙醇+TFA(20︰1)预混合]-甲醇(55︰35︰10),流速0.5 mL·min-1,检测波长278 nm条件下得到部分分离。随后,Aboul-Enein总结了萘羟心安、茚心安、尼比心安在手性色谱柱上的分离,发现这些多手性中心的化合物在基于多糖的手性柱上均能得到分离。1997年,Yaku等[19]在Chiralcel OD和Chiralcel OF柱上成功分离了盐酸地尔硫卓,流动相为正己烷-异丙醇-DEA(19︰1︰0.1)。上述色谱柱多为多糖涂敷型,除此之外还有其他类型的色谱柱应用到多手性中心化合物的手性分析中。2007年,徐贝佳等[20]在自制的替考拉宁柱上[流动相为甲醇-冰醋酸-三乙胺(100︰0.05︰0.10),流速为1.0 mL·min-1,检测波长为225 nm,室温条件]部分分离了拉贝洛尔对映体。2011年,李蓬勃等[21]使用AE Lichrom AM1手性色谱柱直接拆分4种实验室自制的β-氨基酮类双手性中心化合物,在流动相为正己烷-异丙醇二元高压梯度洗脱,流速为1 mL·min-1,检测波长254 nm条件下,4种化合物的4个异构体均达到基线分离。Lisowska-Kuźmicz等[22]用直链淀粉-卵类黏蛋白手性固定相色谱柱手性分离了(±)-反式帕罗西汀,流动相为10 mmol·L-1pH 3.5的磷酸缓冲液-乙腈(98︰2),柱温23 ℃,流速1.5 mL·min-1。Nóra Grecsó等[23]用金鸡纳生物碱手性固定相色谱柱也手性分离了(±)-反式帕罗西汀,并进一步讨论了其手性分离机制。
除了固定相法还有流动相添加剂法,卢铁刚等[24]用C18柱,流动相0.1%磷酸-甲醇(65︰35,含0.38 g·L-1羧甲基-β-环糊精,三乙胺调pH 7.2),柱温25 ℃,210 nm检测波长下基线分离帕罗西汀4个异构体。部分多手性中心化合物在单一的手性柱中只能得到部分分离。Bopp等[25]使用柱切换技术分离了具有双手性中心的对映异构体。柱切换色谱技术又称色谱-色谱联用技术或多维色谱,是利用多通路切换阀切换,改变进样器与色谱柱、色谱柱与色谱柱、色谱柱与检测器之间连接关系的技术。Bopp等使用Chiralcel OA和Chiralpak AD色谱柱切换技术实现了具有2个手性中心化合物4个对映异构体的完全分离,而这是用常规液相色谱法所不能达到的。
图1 尼比心安HPLC手性分离[16]
Fig. 1 HPLC analysis of nebivolol
表1 HPLC分离含多手性中心化合物
Tab. 1 Stereoselective separation of multichiral compounds by HPLC

GC是手性分离中非常重要的色谱方法,关于单手性中心手性分离文献较多,多手性中心手性分离文献较少。1966年,Gil-Av等[26]通过GC实现了首个手性化合物的对映体分离。通常,GC通过手性固定相的氢键、配位和包结作用来进行手性分离,常用的手性试剂主要是氨基酸衍生物、金属配合物、取代基修饰的环糊精等。Armstrong等[27]用环糊精修饰的毛细管柱在气相上分离了反式-1,2-环己二醇,程序升温100~150 ℃,2 ℃·min-1。Schurig等[28]以金属配合物做固定相在GC上分离了具有3个手性中心的2-仲丁基-3-甲基环氧乙烷,其分离柱温为90 ℃。GC目前较多与其他仪器联用(如质谱、液相色谱等),联用后其精确性可以得到进一步的提高。
近年来,CE由于其分析时间短、灵敏度高、流动相用量少且手性试剂选择范围广,已广泛应用于手性分析中,有许多关于CE手性分离的文献[29-31],但是关于多手性中心化合物的手性分析报道较少。CE常用的手性试剂主要有冠醚、环糊精、多糖、大环抗菌药物和配体交换。2004年,Goel等[32]利用毛细管电泳法成功拆分了拉贝洛尔4个对映体,手性试剂为六取代(2,3-二乙酰-6-磺化)-β-环糊精(HDAS-β-CD)和八取代(2,3-二乙酰-6-磺化)-γ-环糊精(ODAS-γ-CD),最佳条件为10 mmol·L-1HDAS-β-CD和10 mmol·L-1ODAS-γ-CD手性试剂溶于低摩尔浓度的pH缓冲液,流动相为25 mmol·L-1磷酸缓冲液(pH 2.5),外加电压为30 KV,检测器波长228 nm,温度25 ℃(图2)。Ana Belén等[33]用1.0%磺化-β-环糊精磷酸缓冲液(pH 2.5)在CE上实现了1种芳香环上有季铵的焦谷氨酸衍生物双手性中心药物的4个对映体手性拆分,且在25 min内达到基线分离。此外,福莫特罗、去甲麻黄碱,顺式-β-内酰胺等在CE上得到部分分离[34-36],CE手性分离技术在多手性中心二肽及三肽类化合物的手性拆分中应用较多,Hammitzsch等[37]用β-CD为手性试剂,分离了二肽类化合物。Salami等[38]用(+)-18-冠-6-四羧酸作手性试剂,分离了氨基酸和一些氨基酸衍生物。除了毛细管电泳,其他毛细管技术如胶束电动色谱、毛细管电色谱等有报道用于多手性中心化合物的分离分析。
胶束电动色谱(MEKC)是由Terabe等人[39]提出的,其手性拆分机制是由于胶束的形成,表面活性剂分子具有2个不同极性的区域,表现出不同特性。表面活性剂有离子型(阳离子和阴离子)、非离子型和两性离子型,这些表面活性剂形成了胶束,对映体的分离就在这些胶束和流动相之间实现。2004年,Rizvi等[40]在实验室合成了手性试剂N-十一烯氧羰基-L-亮氨酸钠(S-L-SUCL)和N-十一烯氧羰基-L-异亮氨酸钠(S-L-SUCIL),对萘羟心安和拉贝洛尔进行对映体拆分,S-L-SUCIL效果比S-L-SUCL更好,用5%磺化-β-CD和S-L-SUCIL做手性试剂,萘羟心安在MECK上4个对映异构体基本达到基线分离,拉贝洛尔能分离得到3个异构体,在25 mmol·L-1S-L-SUCIL和5%磺化-β-CD作为手性试剂时,一次进样可以在35 min内同时分离萘羟心安和拉贝洛尔[41]。
毛细管电色谱(CEC)是1990年开发的介于HPLC和CE之前的混合技术,Mayer等报道了使用开口毛细管CEC进行手性分离[42]。CEC可以在毛细管壁涂覆的开口毛细管柱中进行也可以在不同填料的毛细管柱进行,是色谱机制和电泳机制不同形式的结合。2005年,Bragg等[43]用万古霉素作为手性试剂在CEC上同时分离萘羟心安和拉贝洛尔。结果表明拉贝洛尔4个异构体基线分离,萘羟心安4个异构体中有2个异构体达到基线分离,而另外2个异构体为部分分离。该团队进一步研究了流动相组成、柱温、电场强度等对分离的影响,萘羟心安仍未达到基线分离。
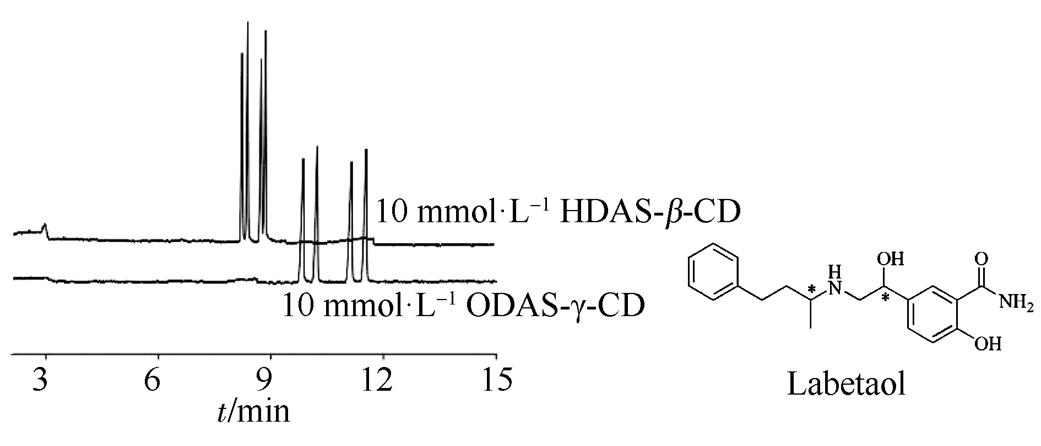
图2 拉贝洛尔CE手性分离[32]
Fig. 2 CE analysis of labetalol
SFC为采用超临界流体作为流动相的色谱方法,最常用的超临界流体是二氧化碳、一氧化二氮、三氟甲烷[44],它与大多数检测器兼容,超临界二氧化碳临界温度和临界压力低,低毒性,环境污染小等优势。1997年,Yaku等成功在Chiralcel OD柱上8 min内分离了地尔硫卓,流动相为二氧化碳-13%异丙醇含0.5%二乙胺的异丙醇,二氧化碳流速为2 mL·min-1,压力180 kg·cm-2,检测器波长254 nm,温度为50 ℃。Lisa等[45]在SFC上采用Chiralpak AD-H柱,以甲醇-乙醇(50﹕50)作为调节剂,直接制备了高纯度的β-甲基苯丙氨酸4个异构体。
CCC是一种连续的不需要固体载体的现代色谱分离技术,相比HPLC,可避免固相吸附带来的污染和样品损失,且进样量大,运行成本低,是手性分离的一种理想的制备色谱技术。CCC由于分离柱效相对较低,在手性分离领域文献报道相对较少。目前,CCC手性分离中已经报道手性选择剂主要有L-脯氨酸衍生物、纤维素与直连淀粉衍生物类、萘普生类、冠醚类、大环抗菌药物类、环糊精类、酒石酸类和金鸡纳生物碱类等。笔者课题组近年来采用CCC已经成功应用并拆分多种外消旋化合物[46]。根据前期基础,基于酒石酸-硼酸配位在CCC上拆分了2个实验室自制的双手性氨基醇类化合物,结构式如图3,这是双手性化合物首次在CCC上成功分离对映体[47]。CCC条件为柱温10 ℃,流速2 mL·min-1,固定相为含0.1 mol·L-1L-酒石酸正己酯的氯仿溶液,流动相为0.1 mol·L-1硼酸溶液(三乙胺调节pH 7.0),进样量为40 mg,保留率60%。化合物(1)由于其对称结构只有3个异构体,经液相色谱检测3个异构体纯度约为98%,化合物(2)有4个异构体,在CCC上分离3个异构体,(+,-)和(-,+)构型在同一逆流峰中,见图3。研究结果表明,只要能筛选到合适的色谱条件,CCC技术也可以应用到多手性中心化合物的分离中。与其他色谱技术相比,CCC具有制备量大、成本低、回收率高等优势。

图3 化合物结构式及CCC手性分离
Fig. 3 Chemical structures of two synthesized compounds and the CCC analysis of compound(1) and compound(2)
多手性中心化合物前期色谱研究主要集中在HPLC上,近年来,CE在手性分离方面逐渐突显优势。CE分析时间短,与HPLC相比,所需样品量少,流动相也只需几毫升,这是其他色谱技术较难做到的一点。多手性中心化合物由于手性中心多,构型复杂,分离条件较单手性中心化合物更难寻找,在HPLC上进行手性分离多采用手性固定相法,流动相组成与配比多元化。目前,在现有条件下,多数多手性化合物不能达到基线分离,还有待进一步研究与探索。
在多手性分离研究方面,开发新型的手性试剂与填料作为固定相或流动相是最为重要的改进措施之一,比如在HPLC、GC、CE、CCC等广泛使用的手性试剂环糊精,探讨更多不同取代基及不同取代度环糊精对手性分离的影响;手性填料也可以互相借鉴,使用在不同的色谱技术上;在较为简单的流动相配比条件下可以进一步尝试更多元化的手性流动相来寻求新的色谱方法;也可以使用色谱柱切换技术或探究新型的色谱形式或色谱组合形式来提高多手性化合物的分离效率。
REFERENCES
[1] ALI I, ALAM S D, AL-OTHMAN Z A, et al. Recent advances in SPE-chiral-HPLC methods for enantiomeric separation of chiral drugs in biological samples [J]. J Chromatogr Sci, 2013, 51(7): 645-654.
[2] ALI I, HUSSAIN A, SALEEM K. Determination of stereo-selective bindings of racemic propranolol with β2-AD-GPCR in human plasma [J]. J Liq Chromatogr Related Technol, 2013, 36(6): 792-806.
[3] CHEN Y Y, LIU T S, LIU R F, et al. Chirotechnology and advances for chiral drugs [J]. J Guangzhou Univ(Nat Sci)(广州大学学报自然科学版), 2002, 1(1): 39-43.
[4] POWELL R J, 王振钺. 药物立体异构体的效能和毒性[J]. 国外医学药学分册, 1989(6): 329-333.
[5] 戴会彬, 王国超, 何高楠, 等. 盐酸地尔硫卓合成研究进展[J]. 化工生产与技术, 2016, 23(2): 31-35.
[6] DALGLIESH C E. The optical resolution of aromatic amino acids on paper chromatograms [J]. J Chem Soc, 1952(137): 3940-3942.
[7] ABOUL-ENEIN H Y. High-performance liquid chromatographic enantioseparation of drugs containing multiple chiral centers on polysaccharide-type chiral stationary phases [J]. J Chromatogr A, 2001, 906(1-2): 185-193.
[8] AL-OTHMAN Z A, AL-WARTHAN A, ALAM S D, et al. Enantio-separation of drugs with multiple chiral centers by chromatography and capillary electrophoresis [J]. Biomed Chromatogr, 2014, 28(11): 1514-1524.
[9] ALI I, SHAIL M, LONE M N, et al. Chiral resolution of multichiral center racemates by different modalities of chromatography [J]. J Liq Chromatogr Related Technol, 2016, 39(9): 435-444.
[10] ALI I, SUHAIL M, AL-OTHMAN Z A, et al. Enantiomeric resolution of multiple chiral centres racemates by capillary electrophoresis [J]. Biomed Chromatogr, 2016, 30(5): 683-694.
[11] 刘建明, 徐媛媛, 陈剑萍. HPLC法在手性药物分离中的应用[J]. 实用临床医学, 2009, 10(2): 137-138.
[12] ABOUL-ENEIN H Y, ALI I. Applications of polysaccharide based chiral stationary phases for resolution of different compound classes [J]. Methods Mol Biol, 2004(243): 183-196.
[13] WANG J Y, DU L M, LI S Q, et al. Determination of enantiomers and diastereoisomers in tadalafil by HPLC [J]. Chin J Mod Appl Pharm(中国现代应用药学), 2017, 34(1): 89-93.
[14] LIU P, ZHANG S Q, ZHANG W S, et al. Determination of chiral impurities inD-hydroxyphenylglycine methyl ester by HPLC [J]. Chin J Mod Appl Pharm(中国现代应用药学), 2017, 34(12): 1731-1733.
[15] MCCARTHY J P. Direct enantiomeric separation of the four stereoisomers of nadolol using normal-phase and reversed-phase high-performance liquid chromatography with Chiralpak AD [J]. J Chromatogr A, 1994, 685(2): 349-355.
[16] DINGENEN J. Polysaccharide phases in enatioseparation. In a practical approach to chiral separations by liquid chromatography [M]. Subramanian G ed. VCH: Weinheim, 1994: 147-150.
[17] ABOUL-ENEIN H Y, SERIGNESE V. Direct HPLC separation of indenolol enantiomers using a cellulose chiral stationary [J]. J Liq Chromatogr Relat Technol, 1996, 19(6): 933-938.
[18] ABOUL-ENEIN H Y, Al-DURAIBI I A. Isocratic HPLC separation of several racemic drugs with two stereogenic centers on a pirkle urea-type chiral stationary phase [J]. J Liq Chromatogr Relat Technol, 1998, 21(12): 1817-1831.
[19] YAKU K, AOE K, NISHIMURA N, et al. Chiral resolution of four optical isomers of diltiazem hydrochloride on Chiralcel columns by packed-column supercritical fluid chromatography [J]. J Chromatogr A, 1997, 785(1-2): 185-193.
[20] 徐贝佳, 张大同, 沈报春, 等. 7种氨基醇类药物在替考拉宁柱上的对映体分离[J]. 分析化学研究报告, 2007, 35(1): 55-60.
[21] LI P B, TIAN Y P, CHEN L R, et al. Resolution of optical isomers of β-amino ketone compounds with two chiral centers by high performance liquid chromatography [J]. Chin J Chrom(色谱), 2011, 29(4): 365-367.
[22] LISOWSKA-KUźmicz M, Kantor-Boruta M, Jończyk A, et al. New validated HPLC methodology for the determination of (−)-trans-paroxetine and its enantiomer in pharmaceutical formulations with use of ovomucoid chiral stationary phase [J]. Anal Bioanal Chem, 2014, 406(15): 3697-3702.
[23] Grecsó N, Kohout M, Carotti A, et al. Mechanistic considerations of enantiorecognition on novel Cinchona alkaloid-based zwitterionic chiral stationary phases from the aspect of the separation of trans-paroxetine enantiomers as model compounds [J]. J Pharm Biomed Anal, 2016(124): 164-173.
[24] LU T G, YANG M J. Separation of paroxetine and its intermediate enantiomers by high performance liquid chromatography using carboxy methyl-β-cyclodextrin as chiral mobile phase additive [J]. Chin J Chrom(色谱), 2007, 25(6): 830-833.
[25] BOPP R J, TACHIBANA K, GEISER F. Pittsburgh Conference and Exposition, March 3-8, 1996 [C]. Chicago: Abstract 448P.
[26] GIL-A V E, FEIBUSH B, CHARLES-SIGLER R. Separation of enantiomers by gas liquid chromatography with an optically active stationary phase [J]. Tetrahedron Lett, 1966, 7(10): 1009-1015.
[27] ARMSTRONG W D, TANG Y B, WARD T, et al. Derivatized cyclodextrins immobilized on fused-silica capillaries for enantiomeric separations via capillary electrophoresis, Gas chromatography, or supercritical fluid chromatography [J]. Anal Chem, 1993, 65(8): 1114-1117.
[28] SCHERIG V, BETSCHINGER F. Metal-mediated enantioselective access to unfunctionalized aliphatic oxiranes: prochiral and chiral recognition [J]. Chem Rev, 1992, 92(5): 873-888.
[29] ALI I, AL-OTHMAN Z A, SALEEM K, et al. Chiral analyses at nano-scale [J]. Comb Chem High Throughput Screen, 2010, 13(6): 562-567.
[30] ALI I, KHAN T A, ABOUL-ENEIN H Y, et al. Chiral analyses of pollutants by capillary electrophoresis [J]. Open Chem Biomed Methods J, 2010, 3(1): 46-55.
[31] ALI I, SANAGI M M, ABOUL-ENEIN H Y. Advances in chiral separations by non-aqueous capillary electrophoresis in pharmaceutical andbiomedical analysis [J]. Electrophoresis, 2014, 35(7): 926-936.
[32] GOEL T V, NIKELLY J G, SIMPSON R C, et al. Chiral separation of labetalol stereoisomers in human plasma by capillary electrophoresis [J]. J Chromatogr A, 2004, 1027(1-2): 213-221.
[33] MARTíNEZ-GIRóN A B, MARINA M L, CREGO A L. Chiral separation of a basic drug with two chiral centers by electrokinetic chromatography for its pharmaceutical development [J]. J Chromatogr A, 2016, 1467(7): 427-435.
[34] CHERKAOUI S, FAUPEL M, FRANCOTTE E. Separation of formoterol enantiomers and detection of zeptomolar amounts by capillary electrophoresis using laser-induced fluorescence [J]. J Chromatogr A, 1995, 715(1): 159-165.
[35] NéMWTH K, VARGA E, IVáNYI R, et al. Separation ofcis-β-lactam enantiomers by capillary electrophoresis using cyclodextrin derivatives [J]. J Pharm Biomed Anal, 2010, 53 (3): 382-388.
[36] LOMSADZE K, VEGA E D, SALGADO A, et al. Separation of enantiomers of norephedrine by capillary electrophoresis using cyclodextrins as chiral selectors: Comparative CE and NMR studies [J]. Electrophoresis, 2012, 33(11): 1637-1647.
[37] HAMMITZSCH-WIEDEMANN M, SCRIBA G K E. Influence of buffer substances and urea on the β-cyclodextrin-mediated chiral separation of dipeptides in CE [J]. Electrophoresis, 2007, 28(15): 2619-2628.
[38] SALAMI M, JIRA T, OTTO H H. Capillary electrophoretic separation of enantiomers of amino acids and amino acid derivatives using crown ether and cyclodextrin [J]. Pharmazie, 2005, 60(3): 181-185.
[39] TERABE S, OTSUKA K, ICHIKAWA K, et al. Electrokinetic separations with micellar solutions and open-tubular capillaries [J]. Anal Chem, 1984, 56(1): 111-113.
[40] RIZVI S A A, SIMONS D N, SHAMSI S A. Polymeric alkenoxy amino acid surfactants: III. Chiral separations of binaphthyl derivatives [J]. Electrophoresis, 2004, 25(4-5): 712-722.
[41] RIZVI S A A, AKBAY C, SHAMSI S A. Polymeric alkenoxy amino acid surfactants: II. Chiral separations of β-blockers with multiple stereogenic centers [J]. Electrophoresis, 2004, 25(6): 853-860.
[42] MAYER S, SCHURIG V. Enantiomer separation by electro chromatography in open tubular columns coated with Chirasil-Dex [J]. J Liq Chromatogr, 1993, 16(4): 915-931.
[43] BRAGG W, NORTON D, SHAMSI S A. Optimized Separation of β-Blockers with multiple chiral centers using capillary electrochromatography–mass spectrometry [J]. J Chromatogr B Anal Technol Biomed Life Sci, 2008, 875(1): 304-316.
[44] SCHOENMAKERS P J. In Packed Column SFC [M]. Smith RM ed. Cambridge: Chromatography Monographs. Royal Society of Chemistry, 1988.
[45] NOGLE L M, MANN C W, JR WATTS W L, et al. Preparative separation and identification of derivatized β-methylphenylalanine enantiomers by chiral SFC, HPLC and NMR for development of new peptide ligand mimetics in drug discovery [J]. J Pharm Biomed, 2006, 40(4): 901-909.
[46] ZHENG Y, YAN J Z, TONG S Q. Research progress on countercurrent chromatography in enantioseparations [J]. Chin J Anal(药物分析杂志), 2013, 33(4): 536-543.
[47] LV L Q, BU Z S, LU M X, et al. Stereoselective separation of β-adrenergic blocking agents containing two chiral centers by countercurrent chromatography [J]. J Chromatogr A, 2017(1513): 235-244.
Progress on Stereoselective Separation of Multichiral Compounds by Chromatography
LYU Liqiong, BU Zhisi, TONG Shengqiang*
(College of Pharmaceutical Science, Zhejiang University of Technology, Hangzhou 310014, China)
ABSTRACT:Drugs with more than one chiral center are usually involved with different stereo-specific isomers and big difference in bioactivity due to different stereo-chemical structure, its separation and analysis is still a great challenge. There are lots of reports about enantioseparation of racemate with one chiral center, however, only a small fraction number of papers about stereoselective separation of racemate with multiple chiral center are available. In this paper, stereoselective separation of racemate with more than one chiral center by various chromatographic techniques, including HPLC, GC, capillary electrophoresis, supercritical fluid chromatography and countercurrent chromatography are reviewed.
KEY WORDS:multichiral centers; chiral analysis; HPLC; capillary electrophoresis
中图分类号:R917
文献标志码:B
文章编号:1007-7693(2018)09-1431-06
DOI:10.13748/j.cnki.issn1007-7693.2018.09.036
引用本文:吕力琼, 步知思, 童胜强. 多手性中心药物色谱拆分研究进展[J]. 中国现代应用药学, 2018, 35(9): 1431-1436.
收稿日期:2017-12-28
作者简介:吕力琼,女,硕士 Tel: (0571)88320984 E-mail: lili6835@126.com
*通信作者:童胜强,男,博士,教授 Tel: (0571)88320984 E-mail: sqtong@zjut.edu.cn
(本文责编:李艳芳)