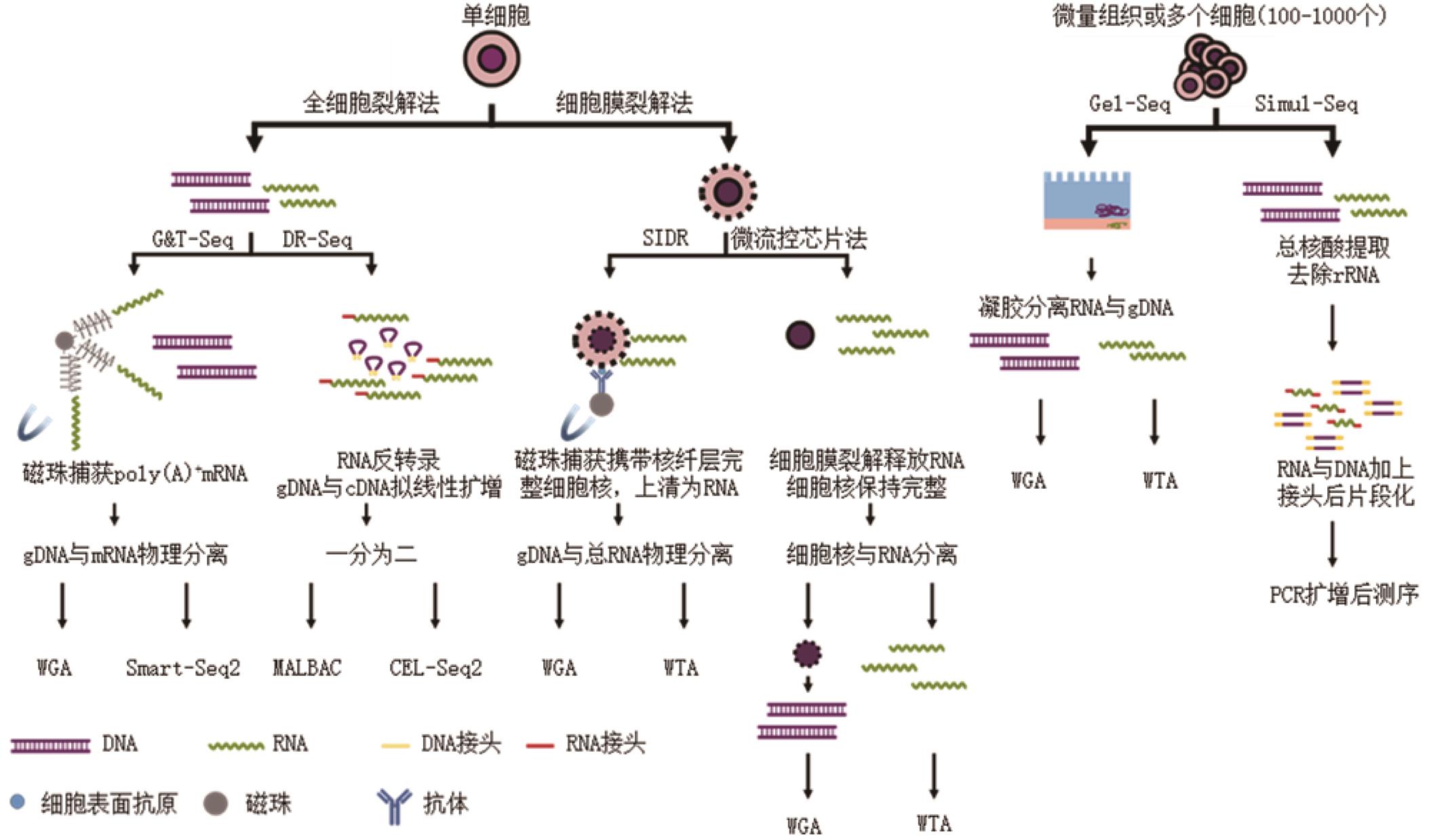
图1 单细胞基因组与转录组共测序方法一览
万睿璇1,周抒遥1,徐欢笛1,汪浩宇1,娄云阁1,姜净1,邹秉杰2,宋沁馨1,2*,周国华1,2
(1.中国药科大学药物质量与安全预警教育部重点实验室,南京 210009;2.南京军区南京总医院药理科,南京 210002)
摘要:单细胞测序提供了大量样品测序所不能提供的异质性等信息。然而由于细胞生长发育受多种因素调节,仅依靠基因型或者表型信息无法全面、准确地说明问题。因此,开发单细胞基因组转录组共测定技术为研究基因变异与表型变异间的关系提供了强有力的工具。目前,单细胞基因组与转录组共测定技术主要分为全细胞裂解法(如:G&T-Seq、DR-Seq),细胞膜裂解法(如:SIDR、微流控芯片法)以及其他方法(如:Simul-Seq、Gel-Seq)。本文对这几类技术进行了简要介绍,并对其前景进行了展望。
关键词:单细胞测序;转录组;基因组;单细胞多组学分析
细胞是构成生命的基本单位。在多细胞生物中,细胞常常受多种因素调节而表现出异质性,这不仅涉及细胞正常发育,也与肿瘤等疾病的发生、发展息息相关[1]。现有的单细胞测序技术可以提供同种或者不同类型细胞的基因组、转录组、表观遗传组,甚至蛋白质组的信息,这使得在单细胞水平上研究正常机体或疾病组织的系统发育、发展成为可能[2-5]。但由于细胞的状态由多种因素共同决定,仅依靠一种分子获得的数据无法准确说明问题,因此,开发整合多组学的技术迫在眉睫。
在这些多组学共测定技术中,基因组与转录组的联合测定十分重要。首先,该技术可关联细胞的基因型与表型信息,实现基因突变对表型影响的定量分析,深入研究导致细胞表型变异的分子原理;另外,应用转录组信息不仅可验证对应细胞DNA的变异,还可进一步了解RNA编辑、等位基因特异性表达对表型多样性产生的影响[6]。值得注意的是,在肿瘤组织中,约有1/3的体细胞编码区单核苷酸突变在RNA有表现,二者同时测序有助于筛选驱动突变(driver mutation)[7]。其次,根据DNA测序结果,利用包含种族信息的DNA结合获得性突变重建细胞的谱系树,并结合同一个细胞的RNA序列信息,进行细胞类型、状态方面的注释,更有助于了解异质性组织的细胞构成与种群结构[8]。因此,基因组与转录组共分析有助于深入理解正常组织和疾病组织之间的遗传学差异,为研究疾病发生机制、进程提供思路[9]。
本文将从以下3个方面对单细胞基因组-转录组共测序技术进行介绍:全细胞裂解法、细胞膜裂解法以及其他可用于微量组织或少量细胞检测的方法,见图1。
图1 单细胞基因组与转录组共测序方法一览
WGA-全基因组扩增;WTA-全转录组扩增。
Fig. 1 Overview of the co-detection of single-cell genome and transcriptome
WGA-whole genome amplification; WTA-whole transcriptome amplification.
该类方法会将整个细胞裂解,核酸会释放至裂解液当中。
2014年,Li等基于多聚胸腺嘧啶肽核酸(oligo-dT peptidyl nucleic acid,PNA)磁珠捕获细胞裂解液中的poly A(+)mRNA,开发了SCTG法(single-cell transcriptogenomics,SCTG)以实现单细胞转录组与基因组的同时分析[10]。但是该方法仅能进行全外显子测序而多用于点突变测定而无法测定基因的拷贝数变异以及结构变异,应用受限。Macaulay等于2015年开发的G&T-Seq (Single Cell Genome and Transcriptome Sequencing,G&T-Seq)通过携带多聚胸腺嘧啶(oligo-dT)引物的磁珠对RNA和DNA进行分离,分离产物可与任意单细胞全转录组扩增(whole transcriptome amplification,WTA)和全基因组扩增(whole genome amplification,WGA)方法联用,且可使用多种测序方法进行分析,无需定制设备,应用范围更广、适用性更强[8,11]。
G&T-Seq方法包括目标细胞的获取、裂解以及DNA与RNA的物理分离[8]。通过流式细胞术(fluorescence-activated cell sorting,FACS)等自动化技术或手动分选获得单个细胞后,将细胞裂解。使用通过链霉亲和素-生物素反应偶联于磁珠表面的多聚胸腺嘧啶引物筛选polyA(+)RNA,此时DNA仍然保留在裂解缓冲液中,实现DNA与RNA的物理分离[11]。随后使用Smart-Seq2(Switching Mechanism at 5’ end of the RNA transcript- Sequencing 2)方法对mRNA进行扩增,并对扩增所得cDNA进行建库测序而获得全长转录组信息[12]。而gDNA可根据分析目的选择适合的全基因组扩增方法,如测定拷贝数变异(CNVs)可以使用多重退火和成环循环扩增(Multiple Annealing and Looping-based Amplification Cycles,MALBAC)以及PicoPLEX,测定单核苷酸变异(SNV)可以使用MDA扩增[8,13-14]。最后对扩增产物进行建库、测序,见图2。
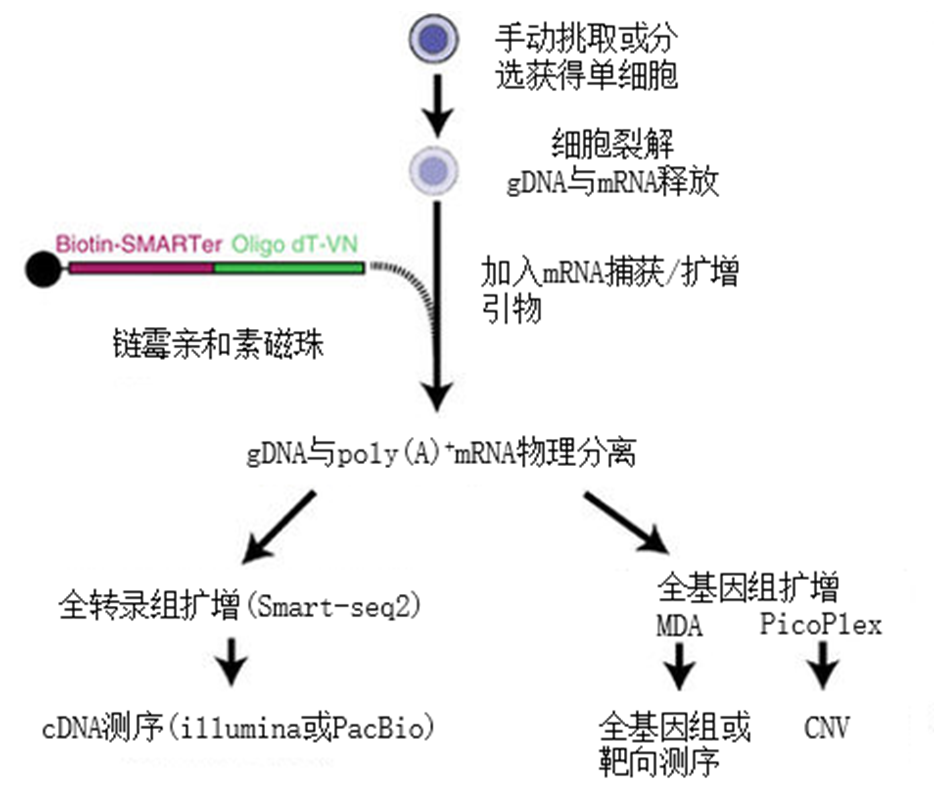
图2 G&T-Seq原理图[8]
Fig. 2 Schematic of G&T-Seq
经G&T-Seq方法物理分离的gDNA与mRNA,可被应用于独立的扩增方法以及测序平台,从而获得基因组与转录组的包括结构变异(Structure Variant)、SNV、CNVs在内的丰富信息。Macaulay等应用G&T-Seq对HCC38乳腺癌细胞进行全长转录本以及基因组分析,确定了造成MTAP-PCDH7融合转录子的染色体重组位置[8]。G&T-Seq还可与Pacific Biosciences RSII或Sequel联用进行cDNA全长测序并提供转录子剪切等信息[11]。使用G&T-Seq分析正常血细胞基因组与转录组后发现极少数细胞11号染色体有3个拷贝,且RNA表达会出现峰值。G&T-Seq用于早期胚胎检测后发现细胞染色体缺失或获得在相应区域的基因表达也会发生降低或上升[8]。另外,经该法分离获得的DNA可用于重亚硫酸盐测序获得甲基化信息[15],结合磁珠捕获而开发的scM&T-seq方法可以用于单细胞的转录组与甲基化的同时测定[16]。
受原理限制,G&T-Seq仅能测定polyA(+)mRNA。但由于选择性多聚腺苷酸化的影响,部分重要基因的polyA长度受到影响,从而影响mRNA捕获[17]。其次,与所有的全基因组扩增、全转录组扩增方法一样,该方法同样会受到扩增歧视的影响而造成覆盖度不均一、等位基因丢失(allelic dropouts,ADO)等假阴性现象,同时受DNA聚合酶错配率的影响产生碱基错配、嵌合DNA分子而造成SNV识别假阳性[13-14,18]。与传统MDA相比,经mRNA分离后的MDA的扩增产物的基因组覆盖分布的均一度有所下降,进一步增加丢失率[8]。另外,对mRNA以及DNA进行开盖物理分离易导致污染的产生。且该法DNA与RNA的物理分离只能手动进行,通量极低,不适用于未来高通量测定的发展方向。
与G&T-Seq不同的是,DR-Seq(gDNA-mRNA Sequencing,DR-Seq)在对DNA和RNA进行共同预扩增后分为2个反应体系进行再扩增。
经人工分选的单个细胞裂解后,释放的mRNA通过经改良的CEL-Seq进行扩增,gDNA使用改良MALBAC法扩增。首先,mRNA在反转录过程中使单链cDNA携带含有T7启动子的Ad-1x接头,并使用携带Ad-2的引物对cDNA以及原有gDNA进行拟线性扩增,随后将样品一分为二,携带Ad-1x和Ad-2的cDNA通过体内转录(in vitrotranscription,IVT)的方式进行扩增,随后进行建库、测序,而仅携带Ad-2的cDNA以及gDNA会通过PCR扩增后建库,进行DNA测序。通过以上步骤,最终实现mRNA、DNA的定量分析[19-22],见图3。
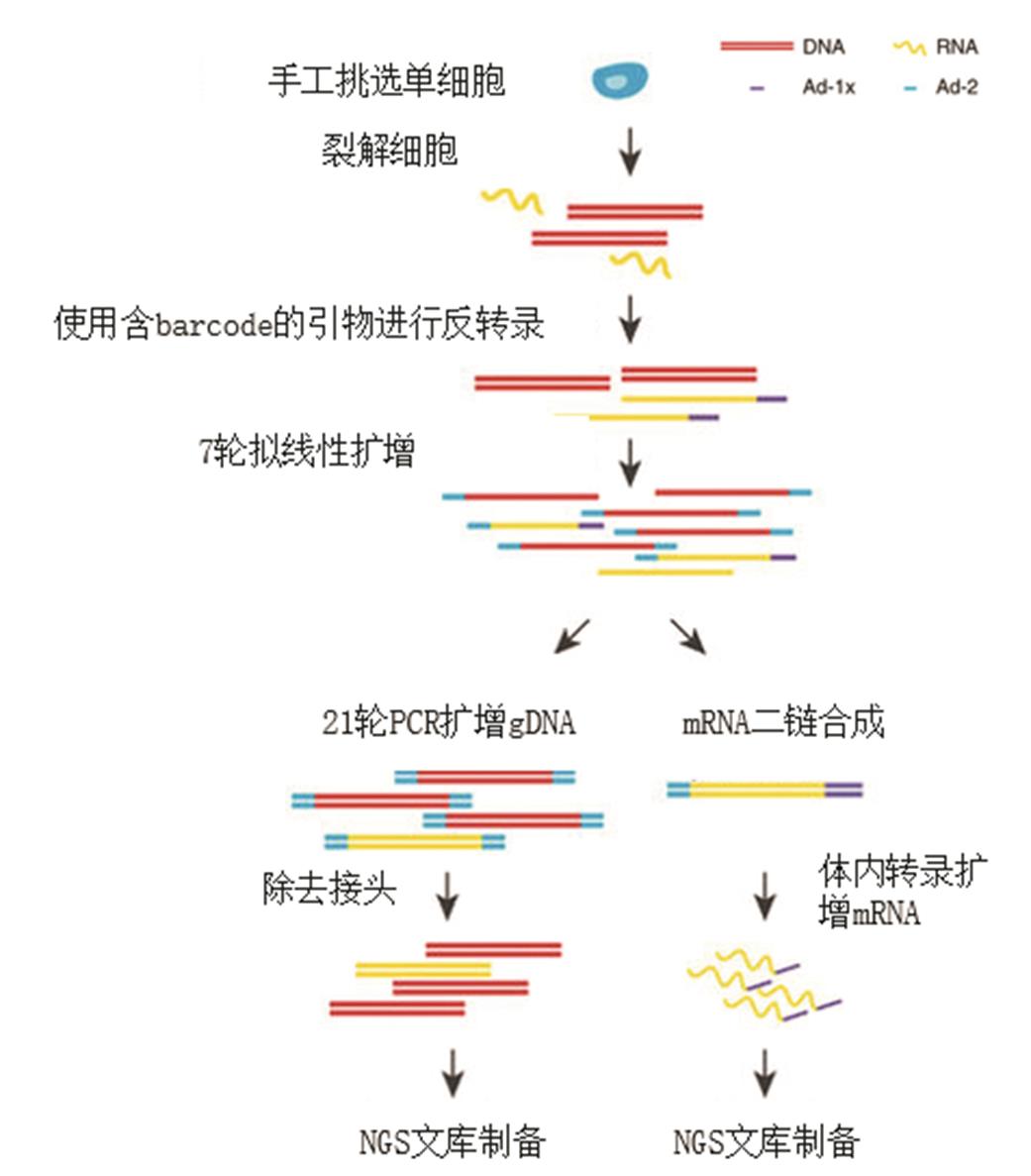
图3 DR-Seq原理图[20]
Fig. 3 Schematic of DR-Seq
Dey等使用DR-Seq对乳腺癌细胞系(SK-BR-3)进行分析后发现在单个细胞乳腺癌细胞中,8号染色体上的基因表达水平正比于基因拷贝数,细胞与细胞间基因表达水平差异与拷贝数发生变化有关[20]。另外,基于小鼠胚胎干细胞的实验证明,DR-Seq还有用于基因组和对应的转录组的SNV的测定的潜力[6,20]。与G&T-Seq相比,由于其为无需物理分离DNA与RNA的单管反应体系,因此不仅减少了由于转移而造成的损失与污染,还可结合微流控平台,提高样品处理通量[11]。另外,Ad-2作为每一个基因的识别条码序列,还可减少扩增歧视,以用于更加准确的基因表达检测。
DR-Seq仅能使用MDA、CEL-Seq分别对DNA和mRNA进行扩增,这大大限制了该方法的应用。受MDA所使用的聚合酶错配率较高的影响,易产生各种假阳性结果[14,23-24]。而CEL-Seq仅富集RNA 3’端,并且更易优先扩增高丰度转录本,无法获取全长cDNA,无法用于转录子融合、剪切变异体等结构变异的研究[11,21],也无法与重亚硫酸盐测序结合获得甲基化信息[6]。由于DNA信息中也包含少量的cDNA信息,导致DNA的拷贝数变异测定时仅能使用内含子序列信息进行分析,这要求预先对待研究基因组有一定了解[8,21,25]。
该类方法在样品制备的过程中仅破裂细胞膜,但是仍然保留完整的细胞核结构。完整细胞核可用于后续的基因组、表观遗传组分析[26-27],细胞质内容物则可用于mRNA分析。
Han等于2017年推出了基于抗体修饰磁珠捕获分离单细胞内DNA与总RNA的SIDR法(simultaneous isolation of genomic DNA and total RNA)[28]。该方法首先使用修饰有抗细胞表面蛋白的抗体(如:Anti-EpCAM)的磁珠捕获目标细胞,随后通过稀释法将细胞分散至48孔板内。之后使用低渗透性缓冲液破坏细胞膜释放RNA,此时核纤层仍然保持完整。随后将裂解物置于磁力架上分离上清与沉淀,上清中包含总RNA,沉淀内容物为gDNA。经该法获得的gDNA可以使用MDA法进行扩增,总RNA可使用SMART-Seq2法进行扩增[29],见图4。
本法提取的DNA与RNA回收率>80%,且DNA与RNA间交叉污染较小,进一步提升了单细胞CNV与SNV识别的准确度。与DR-Seq相比(DNA与RNA无物理分离),经SIDR提取分离的单细胞基因组、转录组具有更高的比对成功率,并且具有更高的覆盖均一度,在染色体范围的CNV识别更具有优势。另外,由于该方法可以提取总RNA,可适用于非编码RNA分析。约90%的基因组会转录成为非编码RNA,但这些非编码RNA在基因表达以及功能方面的作用仍待探索,该方法有望成为研究非编码RNA在细胞中作用的重要工具[30]。
但不可忽视的是,本法通过抗体修饰磁珠对细胞进行捕获结合,极易受到表面抗原而局限于特定细胞种类。同时,本法仅支持手动操作,这也大大影响了分析方法的通量。另外,该法在样品制备以及扩增过程中反应体系仍维持在微升级,与纳升级体系相比,可能会导致扩增歧视发生[31]。
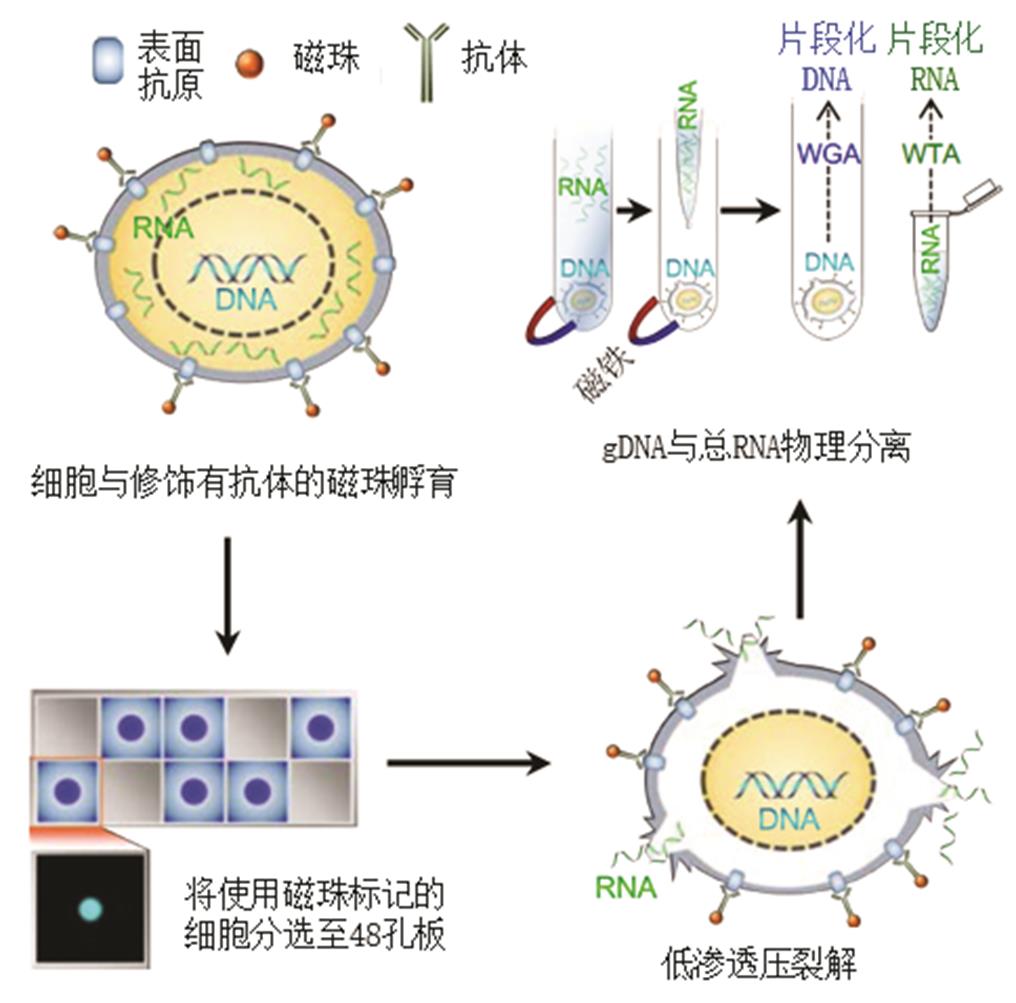
图4 SIDR原理图[28]
Fig. 4 Schematic of SIDR
微流控技术由于具有便于单细胞及其相关组分捕获、处理,省时省力、灵敏度高以及高通量等优势被广泛应用于单细胞分析领域[32-35]。
2014年,Han等首次报道了将微流控平台用于gDNA和mRNA序列的同时测定[36]。该平台使用微流控芯片,在捕获单个细胞后,通过使用特定的裂解缓冲液仅裂解细胞膜的同时保持细胞核完整,随后通过控制芯片上的阀门以分离细胞质内容物与完整细胞核从而实现同一细胞的gDNA和mRNA分离、扩增。随后,结合芯片外PCR、凝胶电泳、Sanger测序等技术即可实现对单细胞的多个转录子及其对应基因的同时分析。
该芯片由流通通道以及控制通道组成。单个细胞首先在微型细胞接种室中捕获,随后使用特殊的细胞裂解液释放细胞质并保持细胞核的完整。细胞质中包含的RNA随后进入芯片上的RNA分支进行第一次反转录,细胞核仍然保留在细胞捕获室中。随后,细胞核在强碱性环境下释放gDNA,并进入芯片上的gDNA分支中变性、中和。紧接着,RNA分支中RNA再次进行反转录与第一次反转录产物连接形成环状cDNA,同gDNA一起进行全池扩增(whole pool amplification,WPA)[27],见图5。
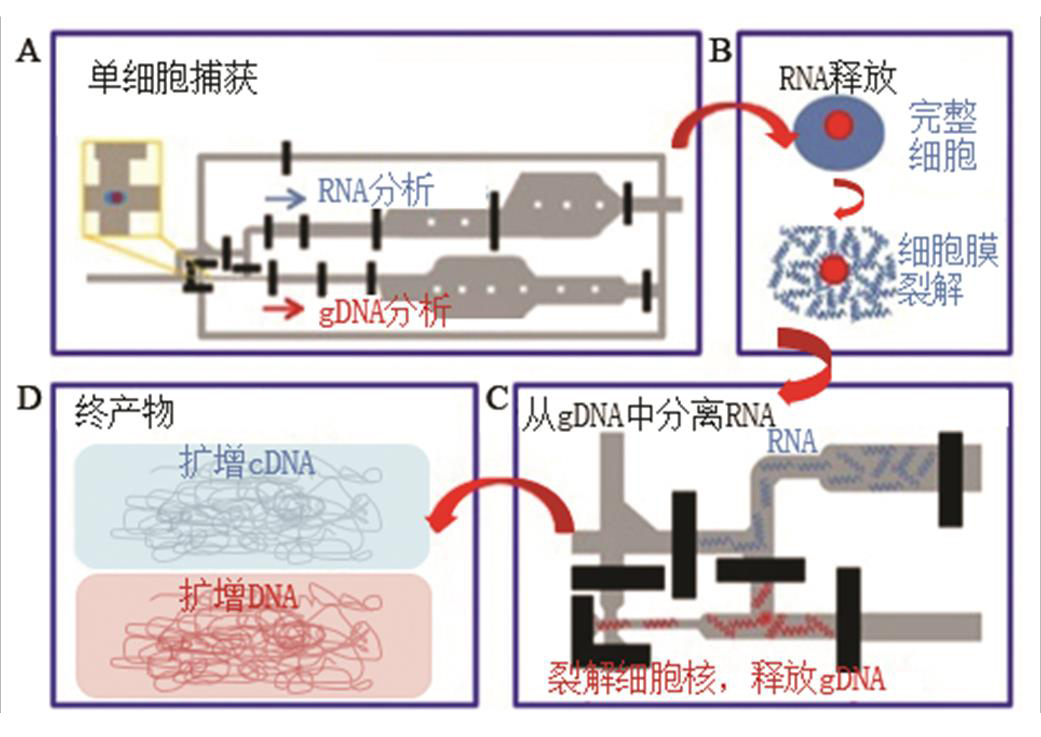
图5 微流控芯片法进行单细胞基因组、转录组共测定原理图[36]
A-单细胞捕获;B-细胞膜裂解释放RNA;C-通过控制阀门使DNA与RNA物理分离;D-RNA反转录为cDNA后扩增,DNA直接扩增
Fig. 5 Schematic of co-detaction of single cell genome and transcriptome via microfluidic platform
A-single cell capture; B-RNA release after cell membrane lysis; C-physical separation of DNA and RNA with the control of valve; D-DNA amplification, RNA reverse transcription and cDNA amplification
该方法最大优势在于除与大规模测序联用外,扩增产物还可与PCR、凝胶电泳、Sanger测序等常规方法联用,以实现同一细胞的基因组与转录组同时定性分析[11]。微流控芯片的使用极大减少了手动操作,并且使用纳升级封闭反应体系,不仅可以减少转移过程中造成的样本损失与污染,还可减少外源DNA以及RNA的污染以及同一细胞内RNA与DNA的交叉污染。体系混合速率提升,避免mRNA的降解,增加反转录的效率。但不可忽视的是,该平台管路由聚二甲基硅氧烷(PDMS)制成,该材料可非特异性吸附DNA或者RNA分子,造成稀有信息的缺失。但该法暂无法实现单细胞全基因组测序或者全转录组测序,仅可进行基因靶向测序[36]。
2017年,Van Strijp等运用相似的思路开发了1个单细胞DNA和RNA共提取微流控平台,并将其用于大肠癌相关驱动通路分析[37]。与上述方法相比,平台不仅由纳升水平升级至皮升水平,并且通过调节压力而非阀门实现液体流动,进一步降低了样品损失以及交叉污染水平,并减少了扩增中可能产生的歧视[31],还可以通过荧光显微镜在处理过程中对细胞进行原位监测。该法可以获得完整的RNA,而不受限于polyA捕获,同时DNA和RNA经物理分离,免去了gDNA序列信息在与基因组比对时需要遮蔽编码区信息的困扰。另外,本平台所得RNA、DNA样本可与现有的商品化单细胞RNA、DNA扩增试剂盒联用,大大提升了方法通用性。经Nextera XT试剂盒建库,样本可用于HiSeq平台进行测序,最终获得更加全面、丰富的基因组与转录组信息。
以下方法未达到单细胞分析水平,但仍然可以分析较低数量的细胞基因组与转录组信息,在CTC细胞团、微量组织异质性研究方面拥有巨大潜力[38]。
Simul-seq的主要分析对象为LCM切割获取的组织样品。
微量组织样品经总核酸提取后,使用RNA 连接酶进行核糖体RNA剔除技术以富集RNA[39]。随后,在Tn5转座酶以及RNA连接酶的作用下,使核酸在片段化的同时添加序列标签(Tagmentation)[40],并在RNA上连接接头。连有接头的RNA进行双链cDNA合成。根据不同的接头对片段进行PCR扩增,之后对扩增片段进行测序[39,41],见图6。
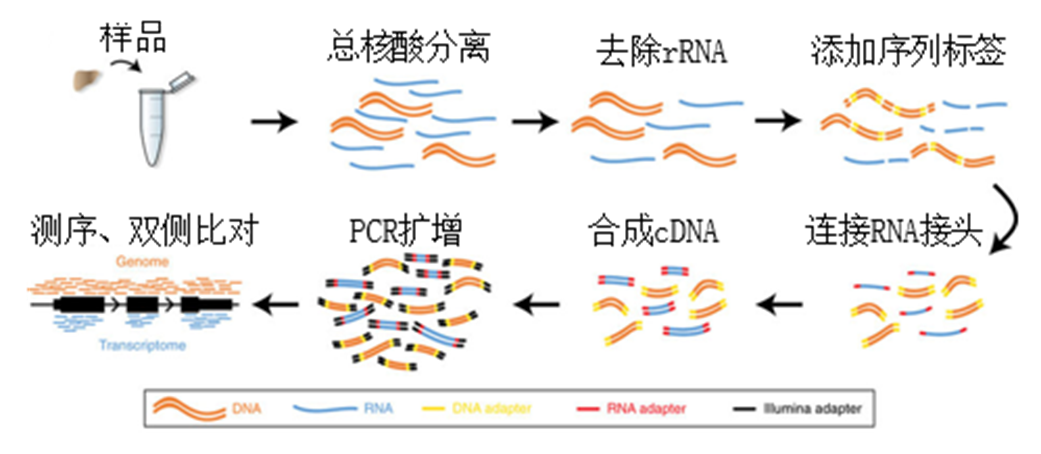
图6 Simul-Seq原理图[39]
Fig. 6 Schematic of Simul-Seq
该法亮点在于可进行非编码RNA的检测。虽然rRNA在总RNA中含量较高,但携带的转录组信息有限,因此使用rRNA去除技术可以大大提高RNA-Seq的效率,同时也保留了mRNA与其余非编码RNA的信息,解决了前述几种方法仅能用于polyA(+)mRNA测定或3’歧视的问题[42]。同时,由于处理后的DNA和RNA所携带的接头不同,大大减少了PCR过程中由于错误连接、标签添加和模板转换产生的交叉污染。与前述几种方法相比,Simul-seq无需对DNA、RNA分别建库,大大减少了文库制备时间,减少了分离过程中可能造成的污染,同时配对数据仍然保持物理连接。Simul-seq为研究肿瘤组织的基因突变、拷贝数变异信息以及非整倍性、对偶基因特异性表现相关信息之间的相互关系提供了可能性。使用Simul-seq对食管腺癌组织进行分析发现非整倍体肿瘤细胞相关基因表达增加,且KIF3B基因突变可能与食管癌发生息息相关。同时,作者还使用该方法筛选了与肿瘤治疗相关的基因[39]。
但是,rRNA删除技术会带来一定的5’歧视,且无法进行单个细胞分析,应用仍然受限[42]。
Gel-seq可以处理包含100~1 000个细胞的样本。
Gel-Seq主要采用不同密度的聚丙烯酰胺凝胶,根据DNA与RNA片段长短,通过电泳将DNA与RNA反转录后得到的cDNA进行分离。经切胶回收,gDNA可直接使用Nextera XT试剂盒建库、测序,而cDNA经PCR扩增后经Smart-Seq处理,可进行建库、测序[25],见图7。
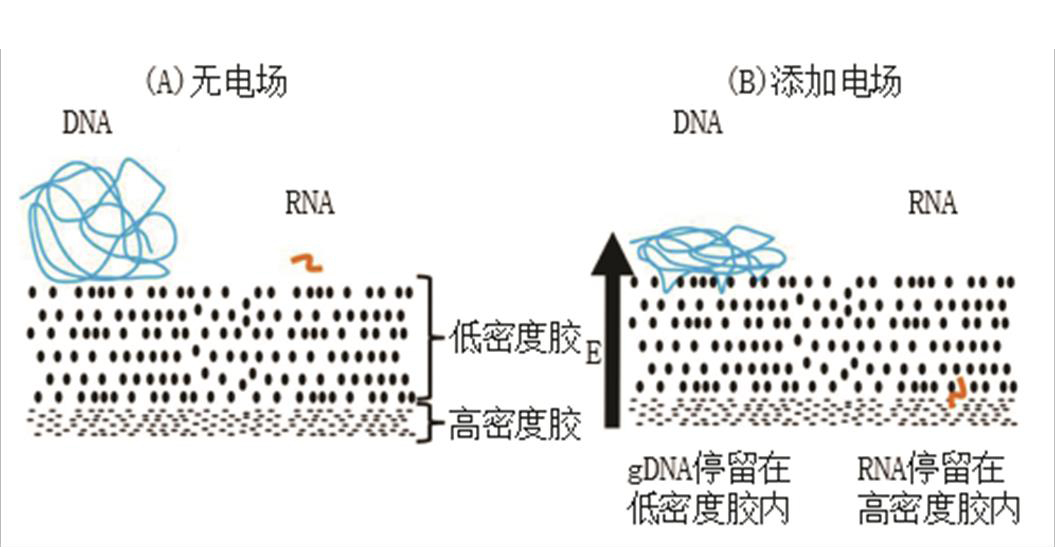
图7 Gel-Seq原理图[43]
A-未施加电场时DNA与RNA状态;B-施加电场时DNA与RNA状态。
Fig. 7 Schematic of Gel-Seq
A-The position of DNA and RNA without electricity field; B-The position of DNA and RNA with electricity field.
Gel-Seq最大的优势在于RNA、DNA处理方法简单,且装置易得,单个样本分析成本较低。但受电泳本身特性的影响,该方法无法用于单个细胞DNA与RNA的分离。另外,在样品处理过程当中有10%~50%的核酸损失率[43]。
由于传统的大规模测序仅能获得样本的平均信息,因此掩盖了较多细胞异质性信息。另外,单一的单细胞基因组测序与转录组测序无法关联同一细胞的基因组与转录组信息,因此单细胞基因组或转录组共测序方法由于可以进行高精度的基因变异与转录子变化关联的分析,应用前景无限。
肿瘤组织、细胞的异质性与肿瘤转移、耐药机制等息息相关。单细胞基因组与转录组共测序技术可以提供详细的肿瘤内基因组、转录组的细胞间差异的图谱,使用共测序技术对肿瘤细胞染色体断点定位、转录子融合以及亚染色体变化的研究,该研究对探寻肿瘤相关基因突变演化进程有极大意义[44]。
除了肿瘤组织外,大脑、肝脏、神经等细胞也存在异质性,不同位置区域的细胞表型与基因型存在较大差异。结合显微组织切割装置(如激光捕获显微切割)获得具有位置信息的单个细胞,可以在不使用表面抗原的情况下对单个细胞进行更准确的分类[45]。除此之外,结合单个细胞的转录组与基因组信息还可研究mRNA翻译与转录爆发等生物现象的原理。
另外,该技术还可用于较难培养的细菌的分析。受限于样本量,此类细菌无法使用常规方法进行分析。而单细胞转录组与基因组共测序可以一次获得细菌的遗传信息,并有可能用于细菌代谢研究,还可应用于耐药菌筛选等[46]。
对单细胞基因组与转录组共测序来说,除了考虑准确度、均一度、覆盖度等基本因素对方法效能的影响,还应考虑样品制备、文库建立过程当中核酸的损失以及核酸分离方法对后续DNA与RNA测序的影响,见表1。在实际研究中,应根据研究对象、目的以及成本等,结合gDNA-Seq与RNA-Seq本质对使用方法进行选择。
目前,单细胞多组学技术发展迅猛,策略多样。Wang等通过将多个单细胞的全基因组、转录组信息结合而获得了慢性淋巴细胞性白血病的驱动基因相关信息[47]。而scTrio-seq可用于单个细胞CNVSs、DNA甲基化、转录组的共同检测[48]。
表1 现有单细胞基因组与转录组共测序方法比较
Tab. 1 Comparision among the methods for the co-detection of single cell genome and transcriptome
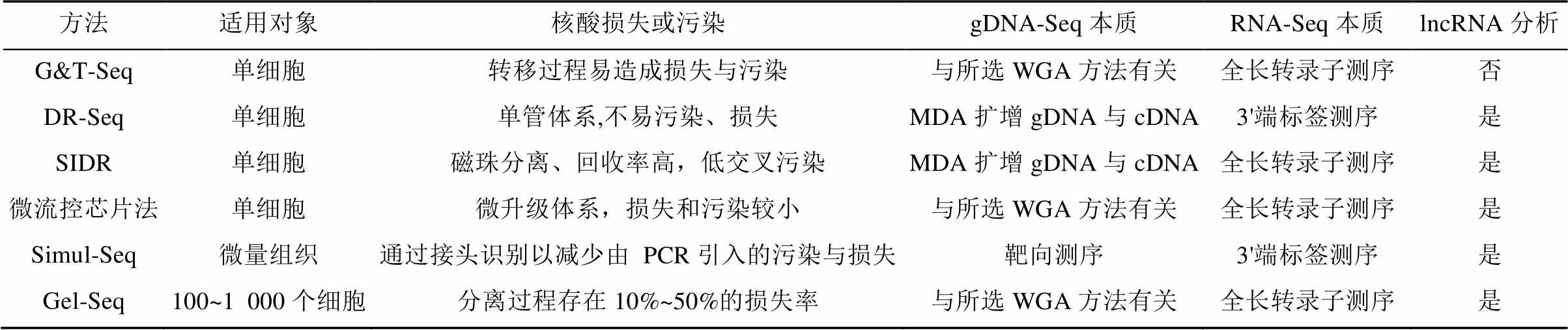
注:lncRNA为长非编码RNA。
Note: lncRNA means long non-conding RNA.
scCOOL-seq可实现单细胞染色质状态、核小体定位、DNA甲基化和基因组的多组学研究[49]。相关研究不能停滞于此。未来,转录组与基因组共测序可以结合空间信息,甚至实现实时检测,并进一步提升基因组测序的准确度和灵敏度以区分真正变异和扩增过程中引入的变异。同时增加反转录效率,最终增加RNA-Seq灵敏度,并扩大RNA检测范围,甚至实现短非编码RNA的测定。另外,微流控芯片由于其自动化、高通量的优势,将会成为未来单细胞多组学研究方法的主要发展方向。
REFERENCES
[1] NICHOLAS N, KENDALL J, TROGE J, et al. Tumor evolution inferred by single cell sequencing [J]. Nature, 2011, 472(7341): 90-94.
[2] GAWAD C, KOH W, QUAKE S R, et al. Single-cell genome sequencing: current state of the science [J]. Nat Rev Genet, 2016, 17(3): 175-188.
[3] ZIEGENHAIN C, VIETH B, PAREKH S, et al. Comparative analysis of single-cell RNA sequencing methods [J]. Mol Cell, 2017, 65(4): 631-643.
[4] SCHWARTZMAN O, TANAY A. Single-cell epigenomics: techniques and emerging applications [J]. Nat Rev Genet, 2015, 16(12): 716-726.
[5] ZHANG R, YUAN H, WANG S, et al. High-throughput single-cell analysis for the proteomic dynamics study of the yeast osmotic stress response [J]. Sci Rep, 2017(7): 42200.
[6] MACAULAY I C, PONTING C P, VOET T. Single-cell multiomics: multiple measurements from single cells [J]. Trends Genet, 2017, 33(2): 155-168.
[7] FUJIMOTO A, TOTOKI Y, ABE T, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. [J]. Nat Genet, 2012, 44(7): 760-764.
[8] MACAULAY I C, HAERTY W, KUMAR P, et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes [J]. Nat Methods, 2015, 12(6): 519-522.
[9] JAFFEE E M, VAN DANG C, AGUS D B, et al. Future cancer research priorities in the USA : a Lancet Oncology Commission [J]. Lancet Oncol, 2017, 18(11): e653-e706.
[10] LI W, CALDER R B, MAR J C, et al. Single-cell transcriptogenomics reveals transcriptional exclusion of ENU-mutated alleles [J]. Mutat Res, 2015(772): 55-62.
[11] MACAULAY I C, TENG M J, HAERTY W, et al. Separation and parallel sequencing of the genomes and transcriptomes of single cells using G&T-seq. [J]. Nat Protoc, 2016, 11(11): 2081-2103.
[12] PICELLI S, FARIDANI O R, BJÖRKLUND A K, et al. Full-length RNA-seq from single cells using Smart-seq2. [J]. Nature Protoc, 2014, 9(1): 171-181.
[13] HOU Y, WU K, SHI X L, et al. Comparison of variations detection between whole-genome amplification methods used in single-cell resequencing. [J]. GigaSci, 2015, 4(1): 37.
[14] HUANG L, MA F, CHAPMAN A, et al. Single-cell whole-genome amplification and sequencing : methodology and applications [J]. Annu Rev Genomics Hum Genet, 2015(16): 79-102.
[15] COKUS S J, FENG S, ZHANG X, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning [J]. Nature, 2008, 452(7184): 215-219.
[16] ANGERMUELLER C, CLARK S J, LEE H J, et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. [J]. Nat Methods, 2016, 13(3): 229-232.
[17] DE KLERK E, AC’T HOENP. Alternative mRNA transcription, processing, and translation: Insights from RNA sequencing [J]. Trends in Genetics, 2015, 31(3): 128-139.
[18] LIANG J, CAI W, SUN Z. Single-cell sequencing technologies: current and future [J]. J Genet Genomics, 2014, 41(10): 513-528.
[19] ZONG C H, LU S J, ALEC R, et al. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell [J]. Science, 2012, 338(6114): 1622-1626.
[20] DEY S S, KESTER L, SPANJAARD B, et al. Integrated genome and transcriptome sequencing of the same cell [J]. Nat Biotechnol, 2015, 33(3): 285-289.
[21] HASHIMSHONY T, WAGNER F, SHER N, et al. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification [J]. Cell Rep, 2012, 2(3): 666-673.
[22] HASHIMSHONY T, NAFTALIE S, GAL A, et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq [J]. Genome Biol, 2016, 17(1): 77.
[23] GRÜN D, VAN OUDENAARDEN A. Design and analysis of single-cell sequencing experiments [J]. Cell, 2015, 163(4): 799-810.
[24] DE BOURCY C F, DE VLAMINCK I, KANBAR J N, et al. A quantitative comparison of single-cell whole genome amplification methods [J]. PLoS ONE, 2014, 9(8): e105585. doi: 10.1371/journal.pone.0105585.
[25] HOOPLE G D. Gel-Seq: an approach for simultaneously sequencing the genome and transcriptome in small populations of cells [D]. Berkeley: University of California, 2016.
[26] HU Y, HUANG K, AN Q, et al. Simultaneous profiling of transcriptome and DNA methylome from a single cell [J]. Genome Biol, 2016, 17(1): 88.
[27] PAN X, URBAN A E, PALEJEV D, et al. A procedure for highly specific, sensitive, and unbiased whole-genome amplification [J]. Proc Natl Acad Sci USA, 2008, 105(40): 15499-15504.
[28] HAN K Y, KIM K T, JOUNG J G, et al. SIDR: simultaneous isolation and parallel sequencing of genomic DNA and total RNA from single cells [J]. Genome Res, 2017, 28(1): 75-87.
[29] PICELLI S, BJÖRKLUND Å K, FARIDANI O R, et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. [J]. Nat Methods, 2013, 10(11): 1096-1098.
[30] HANGAUER M J, VAUGHN I W, MCMANUS M T. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs [J]. PLoS Genet, 2013, 9(6): e1003569. doi: 10.1371/journal.pgen.1003569.
[31] GOLE J, GORE A, RICHARDS A, et al. Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells [J]. Nat Biotechnol, 2013, 31(12): 1126-1132.
[32] PRAKADAN S M, SHALEK A K, WEITZ D A, et al. Scaling by shrinking: empowering single-cell ''omics'' with microfluidic devices [J]. Nat Rev Genet, 2017, 18(6): 345-361.
[33] HOSIC S, MURTHY S K, KOPPES A N, et al. Microfluidic sample preparation for single cell analysis [J]. Anal Chem, 2016, 88(1): 354-380.
[34] KARABACAK N M, SPUHLER PS, FACHIN F, et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples [J]. Nat Protoc, 2014, 9(3): 694-710.
[35] LECAULT V, WHITE A K, SINGHAL A, et al. Microfluidic single cell analysis: from promise to practice [J]. Curr Opin Chem Biol, 2012, 16(3/4): 381-390.
[36] HAN L, ZI X, GARMIRE L X, et al. Co-detection and sequencing of genes and transcripts from the same single cells facilitated by a microfluidics platform [J]. Sci Rep, 2014(4): 6485.
[37] VAN STRIJP D, VULDERS R C M, LARSEN N A, et al. Complete sequence-based pathway analysis by differential on-chip DNA and RNA extraction from a single cell [J]. Sci Rep, 2017, 7(1): 11030.
[38] ACETO N, BARDIA A, MIYAMOTO D T, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis [J]. Cell, 2014, 158(5): 1110-1122.
[39] REUTER J A, SPACEK D V, DAI, P K, et al. Simul-seq: combined DNA and RNA sequencing for whole-genome and transcriptome profiling [J]. Nat Methods, 2016, 13(11): 953-958.
[40] ISLAM S, ZEISEL A, JOOST S, et al. Quantitative single-cell RNA-seq with unique molecular identifiers [J]. Nat Methods, 2014, 11(2): 163-166.
[41] KOZICH J J, WESTCOTT S L, BAXTER N T, et al. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform [J]. Appl Environ Microbiol, 2013, 79(17): 5112-5120.
[42] TARIQ M A, KIM H J, JEJELOWO O, et al. Whole-transcriptome RNAseq analysis from minute amount of total RNA [J]. Nucleic Acids Res, 2011, 39(18): e120.
[43] HOOPLE G D, RICHARDS A, WU Y, et al. Gel-seq: whole-genome and transcriptome sequencing by simultaneous low-input DNA and RNA library preparation using semi-permeable hydrogel barriers [J]. Lab Chip, 2017, 17(15): 2619-2630.
[44] ALIX-PANABIERES,SCHWARZENBACH H, PANTEL K. Circulating tumor cells and circulating tumor DNA [J]. Annu Rev Med, 2012(63): 199-215.
[45] YODA T, HOSOKAWA M, TAKAHASHI K, et al. Site-specific gene expression analysis using an automated tissue micro-dissection punching system [J]. Sci Rep, 2017, 7(1): 4325.
[46] BOCK C, FARLIK M, SHEFFIELD N C. Multi-omics of single cells: strategies and applications [J]. Trends Biotechnol, 2016, 34(8): 605-608.
[47] WANG L, FAN J, FRANCIS J M, et al. Integrated single cell genetic and transcriptional analysis suggests novel private drivers of chronic lymphocytic leukemia [J]. Genome Res, 2017, 27(8): 1300-1311.
[48] HOU Y, GUO H H, CAO C, et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas [J]. Cell Res, 2016, 26(3): 304-319.
[49] GUO F, LI L, LI J Y, et al. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells [J]. Cell Res, 2017, 27(8): 967-988.
Progress in Parallel Sequencing of the Genomes and Transcriptomes of Single Cells
WAN Ruixuan1, ZHOU Shuyao1, XU Huandi1, WANG Haoyu1, LOU Yunge1, JIANG Jing1, ZOU Bingjie2, SONG Qinxin1,2*, ZHOU Guohua1,2
(1.Key Laboratory of Drug Quality Control and Pharmacovigilance of Ministry of Education, China Pharmaceutical University, Nanjing 210009, China; 2.Department of Pharmacology, Nanjing General Hospital, Nanjing 210002, China)
ABSTRACT:Single-cell sequencing provides information, such as heterogeneity, that is omitted by bulk sample sequencing. However, since cell growth and development are regulated by many factors, relying on genotype or phenotype information alone is not able to comprehensively and accurately explain the problem. Co-detection of single-cell DNA-Seq and RNA-Seq offers a strong tool for researchers on the aspect of the relationship between gene variations and phenotype variations. Up to present, there are strategies described below have been employed: whole cell lysis (e.g.: G&T-Seq, DR-Seq), plasma membrane lysis (e.g.: SIDR, microfluidics platform method) and others (e.g.: Simul-Seq, Gel-Seq). This review will introduce and compare these methods in addition to the prediction of their research trends.
KEY WORDS:single-cell sequencing; transcriptomics; genomics; single-cell multiomics
中图分类号:R963
文献标志码:A
文章编号:1007-7693(2018)09-1423-08
DOI:10.13748/j.cnki.issn1007-7693.2018.09.035
引用本文:万睿璇, 周抒遥, 徐欢笛, 等. 单细胞基因组和转录组的共测定方法研究进展[J]. 中国现代应用药学, 2018, 35(9): 1423-1430.
收稿日期:2017-12-24
基金项目:国家自然科学基金项目(81673390);江苏省重点研发计划(社会发展)项目(BE2016745);江苏省基础研究计划(自然科学基金)项目(BK20151445);药物质量与安全预警教育部重点实验室资助项目(DQCP2017MS01);中国药科大学药学基地科研训练及科研能力提高项目(J1310032);江苏省青蓝工程资助项目
作者简介:万睿璇,女,硕士生 Tel: 15298352399 E-mail: ruixuan.wan@stu.cpu.edu.cn
*通信作者:宋沁馨,女,博士,教授 Tel: (025)80860196 E-mail: songqinxin@cpu.edu.cn
(本文责编:李艳芳)