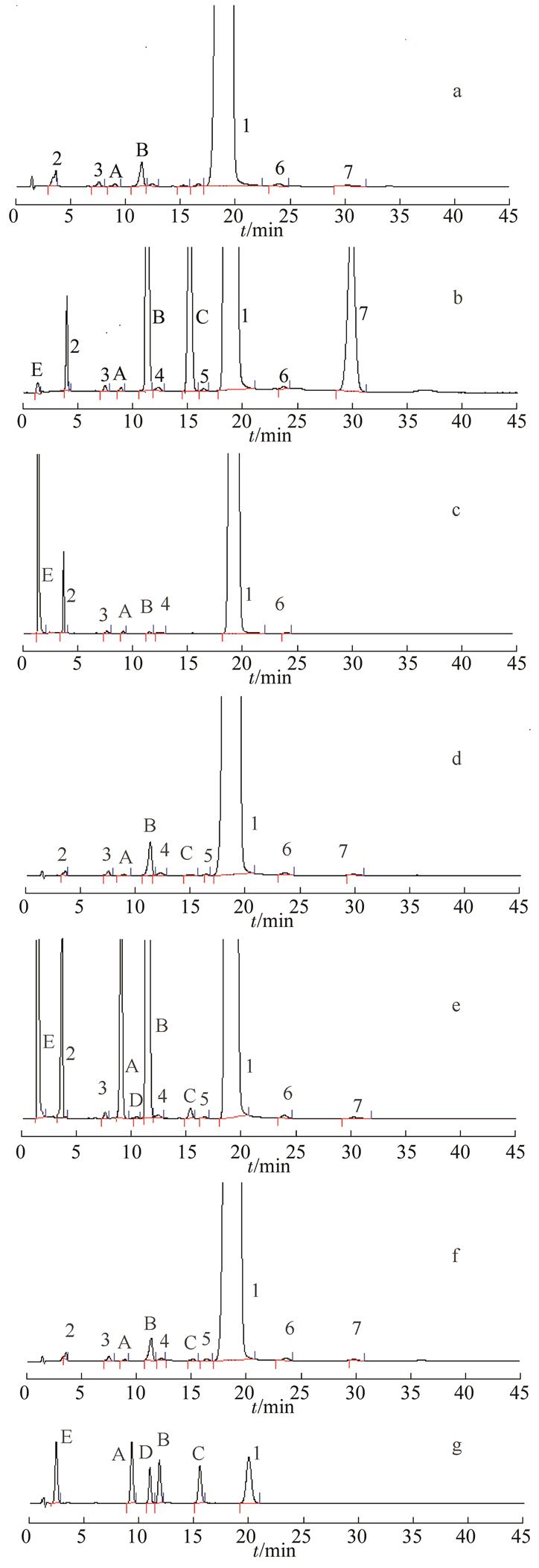
图1 破坏性试验及系统适用性色谱图
孙婷,张西如*,张菁,张轶华
(河北省药品检验研究院,石家庄 050011)
摘要:目的建立保泰松原料及糖衣片剂中5个已知杂质及其他未知杂质检测方法。方法分离条件:Agilent Eclipse XDB-C18(4.6 mm×250 mm,5 μm),柱温为30 ℃,流动相为醋酸铵缓冲液(取醋酸铵2.72 g,加水700 ml溶解,用冰醋酸调节pH值至4.1,加水至1 000 ml,摇匀)-乙腈(58∶42),流速为1.5 mL·min-1,检测波长为254 nm,进样量为20 μl。结果5个已知杂质在25 min内能够完全分离,其中杂质A、B、C、D均在5~30 μg·ml-1,杂质E在0.03~0.15 μg·ml-1具有良好的线性关系;最低检出限量分别为27.86,28.52,26.28,31.96,0.24 ng;原料、糖衣片剂中5个已知杂质的平均回收率分别为98.1%,99.3%,97.6%,97.4%,95.1%和96.9%,97.1%,96.6%,96.1%,94.7%。结论改进的方法灵敏度更高、定量准确,重复性更好,可有效控制保泰松原料及糖衣片剂中杂质含量。
关键词:保泰松;有关物质;高效液相色谱法;杂质;保泰松片;杂质分离;质量控制;方法改进
保泰松(phenylbutazone),又名苯基丁氮酮,属于解热镇痛抗炎药,其作用机制是抑制环氧化酶的活性,从而抑制花生四烯酸最终生成前列环素(PGI1)、前列腺素(PGE1、PGE2)和血栓素A2(TXA2)。临床上常用于治疗类风湿性关节炎、风湿性关节炎及痛风。误服过量或用法不当可引起不良反应,症状为恶心、呕吐、上腹部疼痛、腹泻与便秘[1]。常用的制剂主要是保泰松糖衣片、保泰松丸及保泰松注射液。近期大量研究发现保泰松及其衍生物新的生物活性,例如,刘家萌发现了具有良好的抗HIV活性的4-羟基羟基保泰松[2]。
保泰松原料及糖衣片剂的现行质量标准收载于卫生部药品标准二部第五册[3],均未设置安全性指标有关物质检查项。笔者参阅相关文献[4-12],发现保泰松片有关物质测定方法的文献中,有色谱条件为以乙腈-水(磷酸调pH至3.0)(50∶50)为流动相,检测波长为239 nm[9]。笔者在实际检验中发现,在该色谱条件下无法对收集到的5个已知杂质[10]进行有效分离。本研究对HPLC测定保泰松原料及糖衣片剂中5个已知杂质及未知杂质的色谱柱类型、流动相组成和所选取的pH值进行了比较,优化了试验条件,从而建立了简便的等度洗脱系统分离5个杂质,采用外标法定量,同时也可检测出其他未知杂质,采用自身对照法定量[10]。本实验所建立的方法可使保泰松、5个已知杂质及其他未知杂质得到很好分离,定量准确无干扰,为更好地控制产品质量提供了更有效的检测方法。
Agilent 1260型HPLC仪,包括G1321C四元泵、G1327B自动进样器、G1317A柱温箱、G1314紫外可变波长检测器、Agilent1260 EZChrom化学工作站(美国Agilent公司);XS105型十万分之一电子天平(瑞士梅特勒-托利多公司);MCM36型百万分之一电子天平(Sartorius公司);KQ5200DE超声波清洗器(昆山市超声仪器有限公司)。
保泰松对照品(中国食品药品检定研究院,批号:100481-200601;含量:99.9%,使用前不需干燥);5个杂质对照品来源均为EP;保泰松原料药(不同生产企业提供4批样品);保泰松糖衣片(不同生产企业提供4批样品,规格均为0.1 g);乙腈(色谱纯,Fisher Scientific公司);水为超纯水;冰醋酸(分析纯,天津市风船化学试剂科技有限公司);醋酸钠(分析纯,天津市赢达稀贵化学试剂厂)。
色谱柱为Agilent Eclipse XDB-C18(5 μm,4.6 mm×250 mm);以醋酸铵缓冲液(取醋酸铵2.72 g,加水700 ml溶解,用冰醋酸调节pH值至4.1,加水至1 000 ml,摇匀)-乙腈(58 : 42)为流动相;检测波长为254 nm;柱温为30 ℃;流速为1.5 ml·min-1;进样量为20 μl。理论板数按保泰松计算≥3 000,保泰松峰与其他杂质峰的分离度均符合规定。
精密称取保泰松及5个杂质(杂质A为布马地宗,杂质B为4-羟基保泰松,杂质C为1,2-二苯肼,杂质D为偶氮苯,杂质E为盐酸联苯胺)对照品各适量,加乙腈制成含保泰松25 μg·mL-1和杂质对照品各25 μg·ml-1的溶液。
2.3.1 原料药 精密称取保泰松原料100 mg,乙腈溶解后定容至10 mL量瓶中,即得。
2.3.2 糖衣片剂 取本品,除去包衣,精密称取细粉适量(约相当于保泰松0.1 g),乙腈溶解后定容至10 mL量瓶中,摇匀,滤过,取续滤液,注入液相色谱仪,记录色谱图。
2.3.3 空白辅料 模拟厂家处方,称取空白辅料适量(约1片量),乙腈溶解后定容至10 mL量瓶中,摇匀,滤过,取续滤液,即得。
2.4.1 杂质对照品溶液 精密称取保泰松A、B、C、D杂质对照品各5 mg,乙腈溶解后定容至50 mL量瓶中,作为储备溶液。临用前,取储备液2.5 ml,置10 ml量瓶中,加乙腈稀释至刻度。
2.4.2 杂质E对照品溶液 精密称取保泰松E杂质对照品1 mg,乙腈溶解后定容至100 mL量瓶中,作为储备溶液。精密量取储备液1 ml,置100 ml量瓶中,加乙腈稀释至刻度摇匀,精密量取上述溶液5 ml,置10 ml量瓶中,加乙腈稀释至刻度摇匀,即得。
2.4.3 自身对照溶液 精密量取“2.3”项下制备的供试品溶液l ml,置100 ml量瓶中,用乙腈稀释至刻度,播匀,精密量取上述溶液1 ml,置10 ml量瓶中,用乙腈稀释至刻度,播匀,即得。
吸取空白辅料溶液,系统适用性溶液,对照品溶液及供试品溶液各20 μl,注入液相色谱仪,记录色谱图,按外标法以峰面积计算已知杂质,按自身对照法计算未知杂质。
2.6.1 检出限和定量限 将杂质对照品溶液进行适当稀释后制得一系列不同浓度的溶液,进样分析,以信噪比S/N=3作为检出限,S/N=10作为定量限,A、B、C、D、E检出限分别为27.86,28.52,26.28,31.96,0.24 ng,定量限分别为91.53,94.41,99.61,96.55,0.47 ng。
2.6.2 线性关系考察 配制一系列浓度的A、B、C、D,4个杂质的混合对照品溶液浓度分别为5,10,20,25,30 µg·mL-1;配制一系列浓度的E杂质对照品溶液,浓度分别为0.03,0.04,0.05,0.10,0.15 µg·mL-1。各取20 μL注入液相色谱仪,测定各杂质峰面积,以各杂质对照品浓度为横坐标,色谱峰面积为纵坐标绘制标准曲线,计算5个杂质A,B,C,D,E回归方程分别如下:Y=1.696× 104X-7.390×104,r=0.999 4,Y=1.270×104X-1.523×103,r=0.999 3,Y=1.126×105X+8.333×102,r=0.999 5,Y=8.226×104X-2.272×104,r=0.999 4,Y=1.126× 105X+8.333×102,r=0.999 5,杂质A、杂质B、杂质C、杂质D均在5~30 µg·mL-1内线性关系良好,杂质E在0.03~0.15 µg·mL-1内线性关系良好。
2.6.3 精密度考察 精密吸取杂质对照品溶液20 μL,连续进样6次,测定5个杂质A、B、C、D和E色谱峰峰面积,计算RSD分别为1.3%,1.4%,1.2%,1.6%,1.7%。
2.6.4 专属性试验 该方法可有效分离保泰松原料及其制剂中相关杂质,制剂辅料所带来的相关色谱峰不干扰测定,空白溶剂峰也不干扰测定。该法具有较好的专属性。
2.6.5 破坏性试验
2.6.5.1 未破坏溶液的制备 称取保泰松100 mg,乙腈溶解后定容至10 mL量瓶中,摇匀,作为供试品溶液A。
2.6.5.2 强酸破坏溶液的制备 称取保泰松100 mg,加2 mol·L-1的盐酸溶液1 mL,室温下放置10 min,加2 mol·L-1的氢氧化钠溶液中和至pH值为7.0,再用乙腈定容至10 mL量瓶中,摇匀,作为供试品溶液B。
2.6.5.3 强碱破坏溶液的制备 称取保泰松100 mg,加2 mol·L-1氢氧化钠的溶液1 mL,室温下放置10 min,加2 mol·L-1的盐酸溶液中和至pH值为7.0,再用乙腈定容至10 mL量瓶中,摇匀,作为供试品溶液C。
2.6.5.4 高温破坏溶液的制备 称取保泰松100 mg,加乙腈3 ml溶解,于90 ℃高温烘箱中放置5 h,取出,放置室温,用乙腈定容至10 mL量瓶中,摇匀,作为供试品溶液D。
2.6.5.5 氧破坏溶液的制备 称取保泰松100 mg,加30%过氧化氢溶液2 ml,室温下放置30 min,加乙腈定容至10 mL量瓶中,摇匀,作为供试品溶液E。
2.6.5.6 光破坏溶液的制备 取适量保泰松供试品置培养皿中,在日光下放置5 h后,称取保泰松100 mg,置10 ml量瓶中,加乙腈稀释至刻度,摇匀,作为供试品溶液F。
分别取上述供试品溶液(A~F)20 μL,注入液相色谱仪,记录色谱图。结果显示,未破坏试验(溶液A)图谱中保留时间约11 min有一较大杂质,而在碱破坏试验(溶液C)图谱中该杂质几乎不见,说明该杂质在碱环境中易被破坏降解,转化成其他杂质。结果见图1。
2.6.6 回收率试验 取未含上述5个杂质的保泰松原料及糖衣片剂各9份,每份含主成分约100 mg,精密称定,分别置10 mL量瓶中,精密加入混合杂质对照品溶液(A,B,C,D各250 µg·mL-1,E为0.5 µg·mL-1)0.8,1.0,1.2 mL,每一浓度平行做3份,按“2.3”项下方法制得供试品溶液,分别精密吸取20 μL注入液相色谱仪,测定色谱峰面积,计算回收率。结果见表1~5。
取保泰松原料、糖衣片剂各100 mg,置10 mL量瓶中,分别加乙腈稀释至刻度,摇匀,取得3份供试品溶液。分别测定其室温放置0,0.5,2,4,6,8,10,12 h的主峰峰面积,RSD为0.65%;杂质B及总杂质的峰面积RSD分别为1.3%和1.0%,未有新杂质引入。
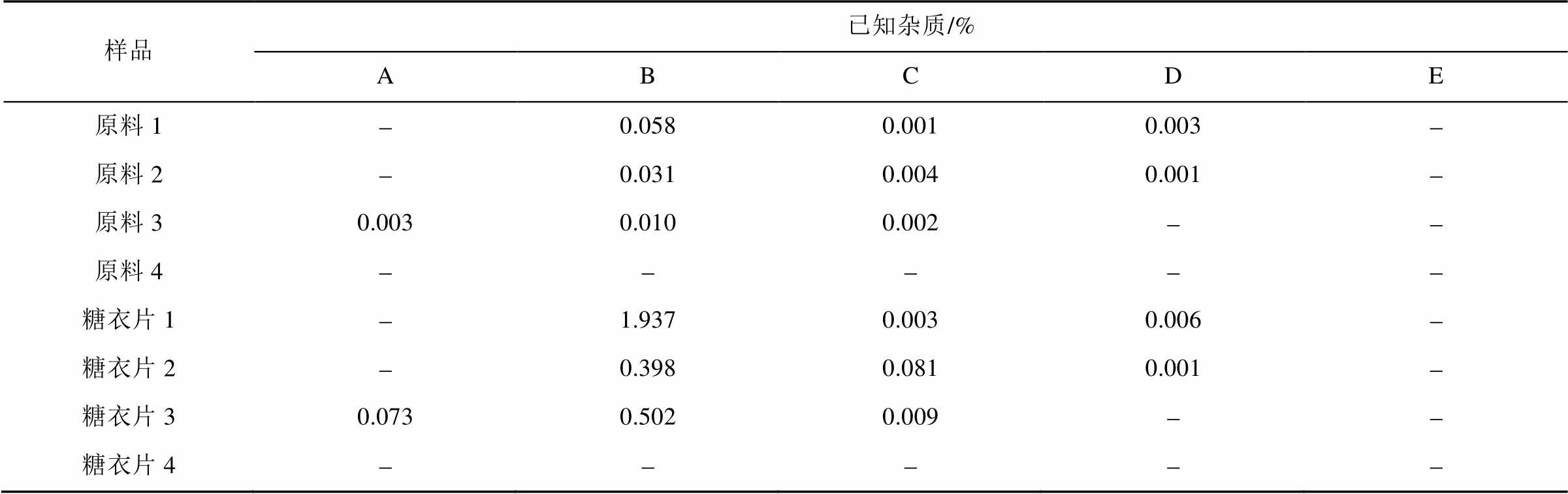
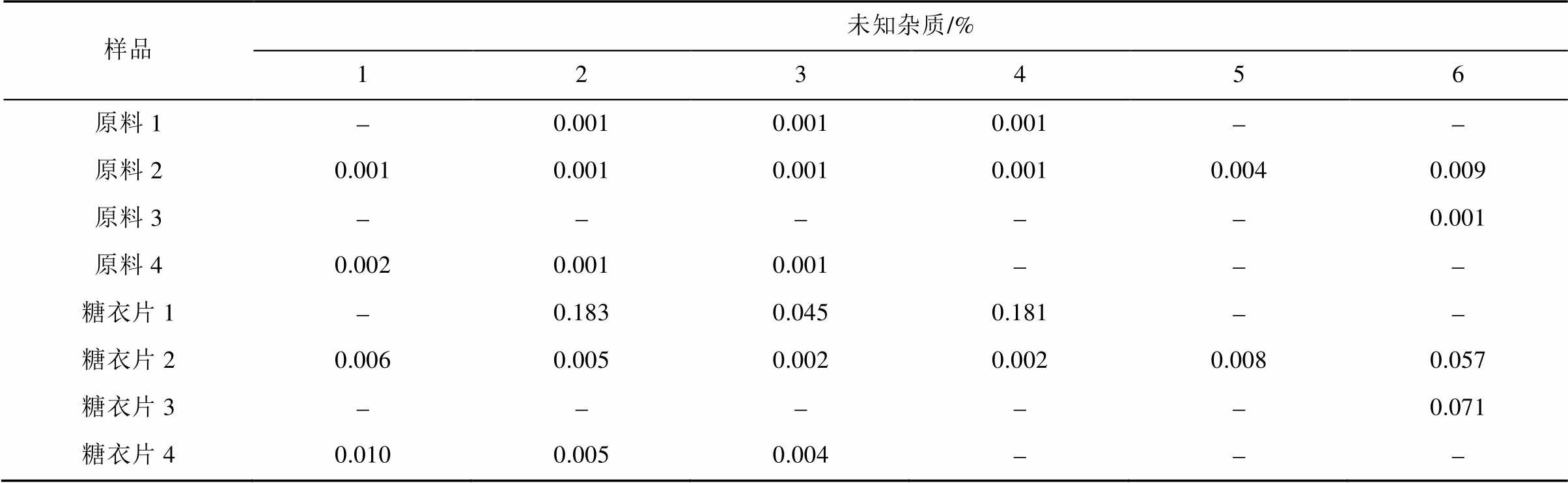
采用面积归一化法,分别按建立的方法测定了不同生产企业提供的保泰松原料、糖衣片剂的有关物质。上述5个杂质在原料和制剂中均未检出杂质E,但有检出其他已知及未知杂质。原料和糖衣片剂各3批的结果见表6~7。
在试验初级阶段采用梯度洗脱方式同时测定样品中的所有杂质,但在系统适用性试验中,已知杂质B和杂质C无法达到有效分离。随之,试验了不同流动相比例的梯度洗脱程序,均未得到很好的改善。最后尝试将梯度洗脱改为等度洗脱,并将pH值调为4.1,并提高流速至1.5 mL·L-1,大大缩短了分析时间,杂质和样品的分离度良好。
图1 破坏性试验及系统适用性色谱图
a-未破坏溶液;b-强酸破坏溶液;c-强碱破坏溶液;d-高温破坏溶液;e-氧化破坏溶液;f-光破坏溶液;g-系统适用性溶液;1-保泰松;2~7为6个未知杂质。
Fig. 1 Chromatogram of system suitability and destructive test
a-undamaged solution; b-sample destroyed by acid; c-sample destroyed by base; d-sample destroyed by heat; e-sample destroyed by oxidation; f-sample destroyed by light; g-system suitability solution; 1-phenylbutazone; 2-7-unkown impurity.
表1 杂质A平均回收率测定结果(n=9)
Tab. 1 The results of impurity A recovery test(n=9)
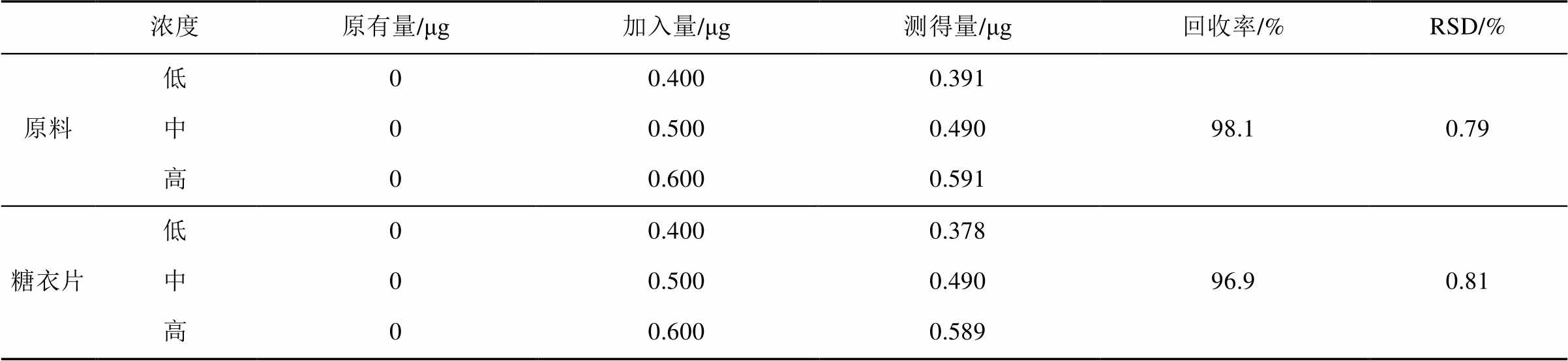
表2 杂质B平均回收率测定结果(n=9)
Tab. 2 The results of impurity B recovery test(n=9)
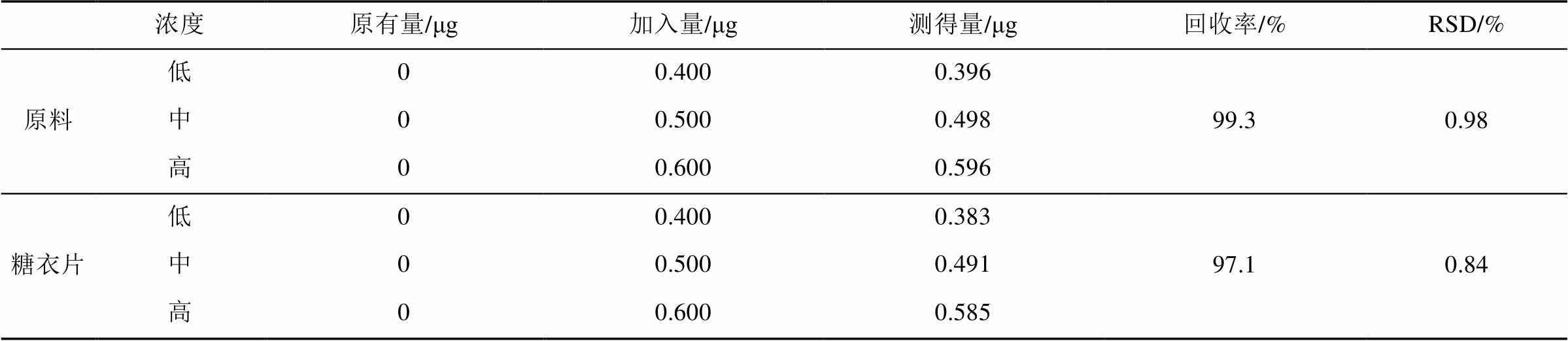
表3 杂质C平均回收率测定结果(n=9)
Tab. 3 The results of impurity C recovery test(n=9)
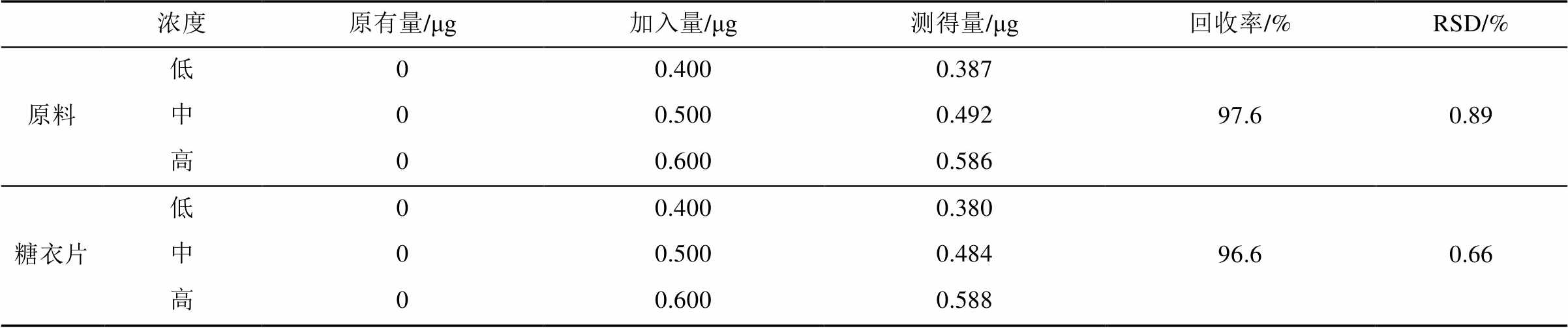
表4 杂质D平均回收率测定结果(n=9)
Tab. 4 The results of impurity D recovery test(n=9)
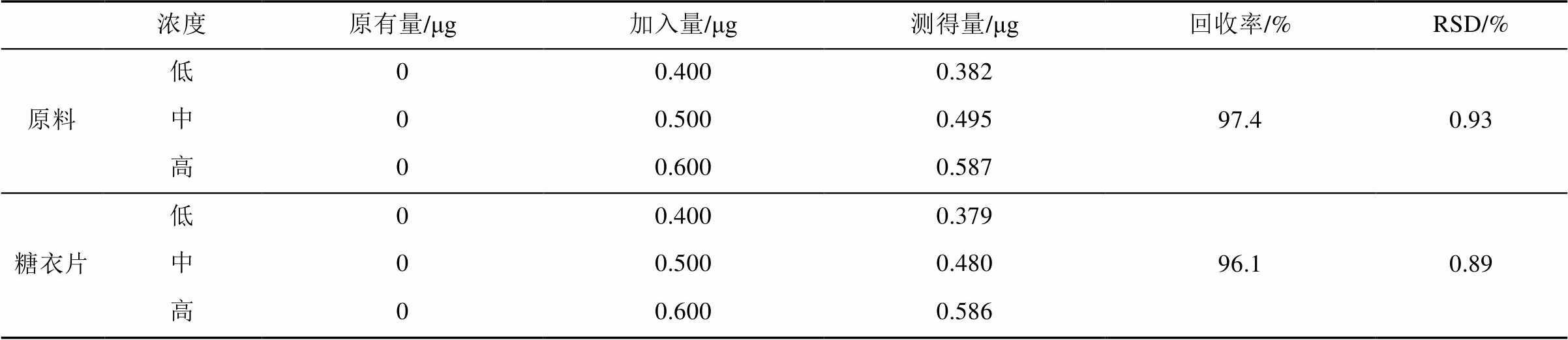
表5 杂质E平均回收率测定结果(n=9)
Tab. 5 The results of impurity E recovery test(n=9)

表6 样品已知杂质测定结果
Tab. 6 The determination results of known related substances
注 :“-”为未检出。
Note : “-” were not detected.
表7 样品未知杂质测定结果
Tab. 7 The determination results of unkown related substances
注:“-”为未检出。
Note: “-” were not detected.
采用紫外检测器检测样品,以EP中梯度洗脱法,检测波长为240 nm(杂质A~D)和280 nm(杂质E)检测发现,该方法色谱柱的耐用性较差,且无法在同一条件下同时检出杂质A~E。本研究发现5个杂质及保泰松对照品在254 nm处检出量均有一定的提高且分离效果良好,故选择254 nm为检测波长。
用5种不同厂家的色谱柱对保泰松片样品进行耐用性试验。结果显示采用5种不同厂家的色谱柱,主峰与已知杂质及未知杂质分离度均>1.5,且按保泰松峰计,理论板数均>4 000,峰纯度良好。表明该方法耐用性良好。
在实际试验过程中发现,杂质C对照品溶液在配制好后,放置6 h后,杂质C会转变为杂质D。因此,杂质C对照品溶液要求临用现配。
曾考虑不去包衣进行试验,在操作过程中发现,该糖衣制剂中糖衣为水溶性,而保泰松为非水溶性,若对未去包衣的样品进行配制,该样品溶液会产生分层现象,不利于试验分析。因此,该实验仍采取去包衣的方式来制备供试品溶液。
随着药品检验技术的发展,方法适用性指标越来越受到人们的关注,在中国药典2015年版二部中,就有很多质量标准在有关物质项下增加了系统适用性试验。越来越多的不良反应事件,也让公众的目光聚焦到药品安全性指标的控制是否完善上。而最能够揭示质量标准是否能对安全性指标进行良好把控的方法便是该方法是否能够检测出已知杂质,且主成分是否能够与已知杂质达到良好的分离效果。因此,体现安全性指标的有关物质检测项,是非常有必要在标准中制定的。并且通过实际检验分析,其中杂质B(4-羟基保泰松)和杂质E(盐酸联苯胺)均为代谢一级毒性物质,需严格把控。本研究在此需求的推动下,改进了保泰松的有关物质的检测方法,能够更加准确、有效地检验保泰松原料及糖衣片剂的已知杂质及未知杂质。为保泰松片质量风险监控提供了有效依据。
保泰松原料及糖衣片剂现行标准中,未设置有关物质检查项,有的文献中虽也提供了可以检查保泰松有关物质的方法,但该方法未检测出保泰松5个已知杂质。通过本研究所采用的方法检测出的保泰松原料及糖衣制剂的有关物质结果可知,该方法可以有效检测出保泰松原料及糖衣制剂的5个已知杂质及未知杂质,且本研究采用的色谱条件具有良好分离效果,能够满足检验需求,且灵敏度高、准确度好。另外,糖衣片剂中杂质B(4-羟基保泰松)检出结果约为0.4~1.9,经对保泰松片生产企业电话咨询,发现不同企业样品采用的原料来源不同,而在保泰松原料生产工艺中需强碱(C2H5ONa)催化,C2H5ONa的生产过程中,不可避免的会带入一部分氢氧化钠,此外,在进行实际存放过程中,考虑到C2H5ONa具有明显的吸水作用,会产生一部分的氢氧化钠,因此对游离的氢氧化钠去除不完全就容易产生副产物杂质B(4-羟基保泰松)。并且在考察影响因素的实验中也发现,杂质B(4-羟基保泰松)在高温的环境中也存在增高的现象。因此推断高温干燥也是杂质B增高的原因之一。通过相关性分析发现,随着贮藏环境温度的升高、相对湿度的增大,杂质B的量都会呈现增长的趋势。严重影响了产品质量,可能会影响用药安全和疗效。
通过以上方法学研究,可证明该色谱条件具有良好的系统适用性,适合测定保泰松片中保泰松原料及制剂有关物质,且该方法还可同时测定保泰松原料及糖衣片剂的含量测定,并且该方法灵敏度高,分离效果及效能好,准确度强,能够较好地控制该制剂的药用质量,为该同类品种药品监督提供有力依据。
REFERENCES
[1] GAO L Y. Determination for the content of the Poltazen with gaseous chromatography [J]. J Shengyang normal univ social Sci Ed(沈阳师范大学学报), 2004, 22(2): 128-130.
[2] LIU J M. Modern prevention and treatment of tuberculosis [J]. Mod prevent med(现代预防医学), 2002, 28(1): 11-14.
[3] Ch.PC Vol ⅡFifth volumes(卫生部药品标准二部,第五册)[S]. 1996: 65.
[4] SUN J W, MA H T. Determination of phenylbutazone tablets by HPLC [J]. Northwest Pharm J(西北药学杂志), 2010, 25(5): 332-333.
[5] LI L, CHEN N J, JIANG Y. HPLC method for content determination of phenylbutazone tablets [J]. Chin J med sci(中国医药科学), 2015, 5(17): 61-63.
[6] ZHANG L, BAI Q S, WANG L. Content determination and quality control of compound belladonna oral solution [J]. J pharm anal(药物分析杂志), 2013, 33(8): 1407-1410.
[7] MU T N, WANG H, LIU Y Q, et al. Determination of phenylbutazone in cosmetics by high performance liquid chromatography [J]. Daily chem indust(日用化学工业), 2011, 41(6): 459-461.
[8] 杜兴. HPLC法测定风湿松片中保泰松及氨基比林的含量 [J]. 西北药学杂志, 1999, 14(2): 53.
[9] LI L, JIANG Y, CHEN N J. HPLC determination of related substances of phenylbutazone tablets [J]. China Med Pharm (中国医药科学), 2015, 5(19): 72-74.
[10] WANG X L, LIU X F, XI Z F, et al. Construction of a determination method for the related substance in crude drug and preparation of miconazole nitrate [J]. J pharm anal(药物分析杂志), 2012, 32(11): 2025-2030.
[11] ZHAO J L, ZHANG M, ZHANG X R, et al. Improvement on determination of hydroquinone and phenol inwhitening cosmetics by HPLC [J]. J pharm anal(药物分析杂志), 2017, 37(3): 508-513.
[12] CHE H Y, YANG Y Y, FENG A P. Determination of phenylbutazone and chlorphenamine maleate in arthrosis analgestics tablets by HPLC [J]. Chin Pharm Affa(中国药事), 2003, 17(3): 167-168.
(本文责编:蔡珊珊)
Improvement Research of a determination method for the related substance in crude and preparation of phenylbutazone by HPLC
Sunting, Zhangxiru*, Zhangjing, Zhangyihua
(Hebei institute for drug control, shijiazhuang 050011, China)
ABSTRACT: OBJECTIVETo establish a method to detect five kinds of known impurities and others unknown impurities in phenylbutazone crude drug and coated tablet.METHODSThe HPLC analysis was performed on a Agilent Eclipse XDB-C18(5 μm, 4.6 mm×250 mm)column; the column temperature was 30 ℃; the mobile phase consisted of ammonium acetate buffer (take ammonium acetate 2.72 g, dissolve 700 ml of water, adjust the pH to 4.1 with glacial acetic acid, add water to 1 000 ml, shake well) and acetonitrile(58∶42) at the flow rate of 1.5 mL·min-1, the detection wavelength was 254 nm, and injection volume was 20 μl.RESULTSFive kinds of impurities were completely separated in 25 min, and good linear relationships of A, B, C, D were obtained in the range of 5-30 μg·ml-1. good linear relationships of E was obtained in the range of 0.03-0.15 μg·ml-1, the limit of detection of were respectively 27.86, 28.52, 26.28, 31.96, 0.24 ng; the average recoveries were respectively 98.1%, 99.3%, 97.6%, 97.4%, 95.1% and 96.9%, 97.1%, 96.6%, 96.1%, 94.7%.CONCLUSIONThe improved method is more sensitive and reproducible, and can be used for quality control of the crude drug and preparation of phenylbutazone efficiently.
KEY WORDS:phenylbutazone; related substance; HPLC; impurity; phenylbutazone tablets; impurities separation; quality control; method improvement
中图分类号:R917.101
文献标志码:B
文章编号:1007-7693(2018)03-0357-06
DOI:10.13748/j.cnki.issn1007-7693.2018.03.011
引用本文:孙婷, 张西如, 张菁, 等. HPLC测定保泰松原料及糖衣片剂有关物质的改进研究[J]. 中国现代应用药学, 2018, 35(3): 357-362.
收稿日期:2017-08-01
基金项目:河北省科技计划项目(162777106D)
作者简介:孙婷,女,硕士,主管药师 Tel: (0311)85212008 E-mail: 122547652@qq.com
*通信作者:张西如,女,主任药师 Tel: (0311)85212008 E-mail: meshall1983@126.com