图1 化合物1~3的化学结构
Fig. 1 Structures of compounds 1-3
谭娟,陈灵,彭安林,肖静,张恩景
(武汉大学附属同仁医院 武汉市第三医院药学部,武汉 430060)
摘要:目的设计、合成杂环二茂铁衍生物,并研究其抗三阴性乳腺癌活性。方法以二茂铁查耳酮为先导化合物,对其进行结构改造,合成了一系列含有杂环的二茂铁衍生物,并通过CCK8试剂盒测试化合物抗乳腺癌活性。结果合成了28个二茂铁衍生物,其结构均通过1H-NMR和MS加以确证。初步的生物活性测试结果表明,所合成的二茂铁衍生物对三阴性乳腺癌MDA-MB-231细胞有较强的选择性和抑制活性,其中咪唑杂环化合物抗肿瘤活性强于相应的吡唑类和嘧啶化合物。尤其是28a[IC50=(1.6±0.23)μmol·L-1]对MDA-MB-231的抑制活性分别是先导化合物3[IC50=(10.7±1.41)μmol·L-1]和他莫昔芬[IC50=(13.7±1.17)μmol·L-1]的6和10倍,同时这些二茂铁衍生物对正常乳腺上皮细胞MCF-10A均没有毒性。结论本研究为开发具有抗三阴性乳腺癌活性的化合物提供了信息和依据。
关键词:二茂铁衍生物;合成;抗三阴性乳腺癌
三阴性乳腺癌(triple negative breast cancer,TNBC)是乳腺癌中恶性程度较高的类型,约占乳腺癌病理类型的10%~20%[1]。在治疗上由于缺乏TNBC有效的特异性靶点,因此传统的靶向药物对之无效。对TNBC来说,其复发率和转移率特别高,这也是该种类型乳腺癌有较高死亡率的原因[2]。临床上对于TNBC的治疗,主要是以铂类、蒽环类、紫杉醇类为基础的化疗[3],但其全身药物的不良反应严重,药物毒性大,严重影响了患者的生活质量。因此,开发出新型、高效、低毒的抗TNBC药物已成为医疗界迫切的需要,同时也是当今学术界和制药界的研究热点[4]。
近年来研究发现,查耳酮作为一类重要的天然产物也表现出了抗乳腺癌活性[5],同时对正常细胞没有毒性。本课题组设计、合成了一系列的查耳酮,并对TNBC展现出了中等的抑制活性(图1,化合物1,IC50>29mmol·L-1)[6];随后,笔者对该类查耳酮化合物进行结构修饰,引入二茂铁(图1,化合物2)基团构建了一系列的二茂铁查耳酮,显著地提高对TNBC的抑制活性,其中化合物3 [图1,IC50=(10.7±1.41)mmol·L-1]展现出了最强活性[7],但与他莫昔芬相比,大部分二茂铁类查耳酮抗乳腺癌活性仍然较低。因此,拟进一步对二茂铁查耳酮进行结构优化,以期望得到抗TNBC活性更强、毒性更低的新型二茂铁查耳酮衍生物。
参考查耳酮的结构改造经验发现,将查耳酮a,b-不饱和酮修饰成杂环后能提高抗肿瘤活性[8-11]。因此,笔者拟构建一系列杂环类二茂铁衍生物,其合成过程见图2~3。首先,以1-二茂铁基-3-(4-氯苯基)-2-丙烯酮(化合物3)为先导化合物,将其修饰成嘧啶(化合物4~10)杂环后能够显著提高其抗乳腺癌活性;接着,将化合物3修饰成二氢吡咯杂环化合物11~17后,其抗TNBC活性较嘧啶类化合物进一步提高,其中N-1位为4-F苯基取代的化合物17a活性强于其他取代基的化合物,而将化合物17a的二氢吡咯环氧化成吡咯环(化合物18)后却降低了抗肿瘤活性;随后,保留了N-1位上4-F苯基,合成了4-醛基二茂铁化合物21,并通过Claisen-Schmidt羟醛缩合反应合成了另一系列具有咪唑杂环的二茂铁类查耳酮22~28。通过生物活性研究发现,这类咪唑杂环化合物对TNBC的抑制活性强于二氢吡咯杂环类,其中化合物28a[IC50=(1.6±0.23)mmol·L-1]展现出了最强的抑制活性,是先导化合物3的6倍。
图1 化合物1~3的化学结构
Fig. 1 Structures of compounds 1-3
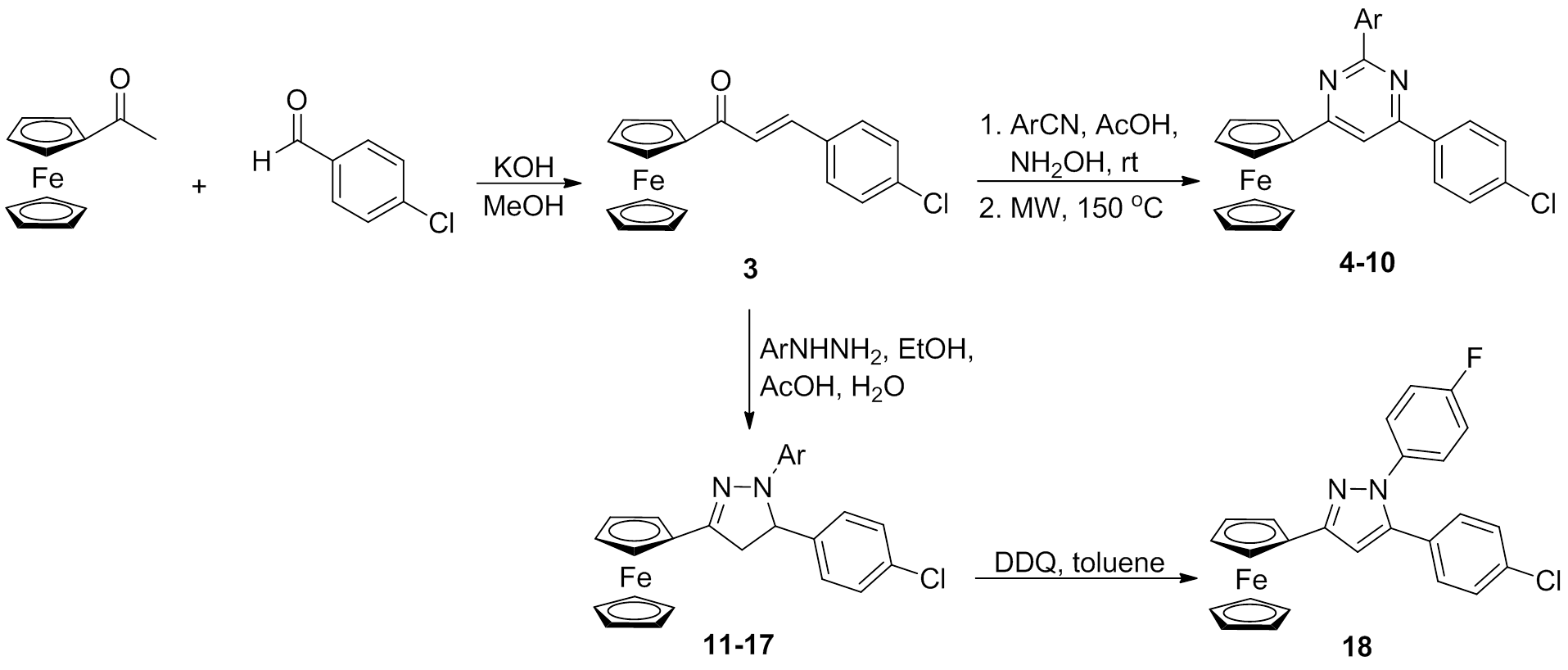
图2 二茂铁类化合物4~18的合成路线
Fig. 2 Synthetic route of ferrocenyl compounds 4-18

图3 二茂铁类化合物22~28的合成路线
Fig. 3 Synthetic route of ferrocenyl compounds 22-28
BrukerAM-400Hz型核磁共振仪(TMS内标);XR4显微熔点测定仪(上海光学仪器厂);DF-101S 集热式恒温加热磁力搅拌器(河南巩义市予华仪器有限责任公司);FA(N)/JA(N)系列MINQI∧O电子天平(上海民桥精密科学仪器有限公司);1106型元素分析仪(意大利Carlo Erba公司);HP1100质谱仪(美国Agilent公司);GF254硅胶薄层板(青岛海洋化工厂)。
乙酰基二茂铁(北京百灵威科技有限公司,纯度>98%);对氯苯甲醛(北京百灵威科技有限公司,纯度>98%);苯肼衍生物(阿拉丁试剂有限公司,纯度>95%);苯甲氰衍生物(阿拉丁试剂有限公司,纯度>95%);合成使用的其他试剂均为分析纯;水为蒸馏水。
称取单乙酰基二茂铁(4.347 g,12.4 mmol)和对氯苯甲醛(1.736 g,12.4 mmol)溶解到50 mL甲醇中,将反应瓶置于冰浴中,冷却后缓慢加入KOH (3.484 g,62.1 mmol),撤除冰浴后在室温下反应12 h,用10%盐酸调节溶液pH 3,析出红色沉淀,过滤,滤饼在真空干燥箱中干燥过夜即得到化合物3,产率:78%,m.p. 158~160 ℃;1H-NMR (400 MHz,CDCl3)δ7.64 (d,J= 15.6 Hz,1H),7.49 (d,J= 8.4 Hz,2H),7.30 (d,J= 8.4 Hz,2H),6.99 (d,J= 16.0 Hz,1H),4.83 (s,2H),4.52 (s,2H),4.13 (s,5H)。
称取二茂铁查耳酮(化合物3,0.421 g,1.2 mmol)、苯甲氰衍生物(1.2 mmol)、羟胺(0.041 g,1.2 mmol)溶解到5 mL醋酸中,室温反应2.5 h后,将反应液在微波(600 W,180℃)中反应5 min,将反应液冷却至室温,加入15 mL饱和的NaCl溶液,用乙酸乙酯萃取(3×30 mL),有机层用无水硫酸钠干燥,减压除去溶剂,经柱层析(石油醚-乙酸乙酯,4∶1)得到相应的嘧啶类化合物4~10,其收率、理化常数及波谱数据见表1~2。
氩气保护下,将二茂铁查耳酮(化合物3,0.386 g,1.1 mmol)和苯肼衍生物(1.1 mmol)溶解到无乙醇(10 mL)、醋酸(6 mL)与水(2 mL)的混合溶剂中,加入回流反应6 h,将反应液冷却至室温,倒入20 mL冰水中,析出红色的沉淀,过滤,滤饼真空干燥,甲醇重结晶得到相应的二氢吡唑类化合物11~17,其收率、理化常数及波谱数据见表1~2。
称取化合物17a(0.279 g,0.6 mmol)和二氯二氰基苯醌(DDQ,0.091 g,0.4 mmol)溶解到苯(20 mL)中,加热至回流反应4 h,将反应液冷却至室温,减压除去溶剂,经柱层析(石油醚-乙酸乙酯,2∶1)得到吡唑化合物18,其收率、理化常数及波谱数据见表1~2。
氩气保护下,称取乙酰二茂铁(化合物1,0.386 g,1.1 mmol)和4-氟苯肼(1.1 mmol)溶解到无乙醇(20 mL)中,缓慢滴加1 mL浓硫酸,室温反应12 h,用10% NaOH溶液调节pH 8,析出红色的沉淀,过滤,滤饼真空干燥,甲醇重结晶得到化合物20,产率为84%,m.p. 184~186 ℃;1H-NMR (400 MHz,CDCl3)δ7.53 (d,J= 8.4 Hz,2H),7.29 (d,J= 8.0 Hz,2H),7.08 (d,J= 8.0 Hz,2H),6.88 (d,J= 8.4 Hz,2H),4.72 (s,2H),4.22 (s,5H),4.11 (s,2H),2.97 (s,3H)。
冰浴下,称取化合物19(0.386 g,4.1 mmol)溶解到POCl3(0.5 mL,6.1 mmol)和DMF(10 mL)的溶液中,加热至60 ℃反应6 h,反应液冷却至室温后倒入50 mL的冰水中,用10% NaOH溶液调节pH 7,析出红色的沉淀,过滤,滤饼真空干燥,甲醇重结晶得到化合物21,产率: 71%,m.p. 169~172 ℃;1H-NMR (400 MHz,CDCl3)δ7.61 (d,J= 8.0 Hz,2H),7.28 (d,J= 8.0 Hz,2H),6.84 (s,1H),4.71 (s,2H),4.24 (s,5H),4.13 (s,2H)。
称取化合物20(0.598 g,2.3 mmol)和相应的芳乙酮(1.6 mmol)溶于20 mL无水甲醇中,室温搅拌10 min后,将反应液置于冰浴中冷却,缓慢加入10 mL 20%的KOH溶液,反应液呈红色。在室温下继续反应12 h,用20%的盐酸调节pH 3后,部分产物以沉淀析出后过滤,滤饼在真空干燥箱中干燥;如加盐酸调节pH后未能析出沉淀则用乙酸乙酯(3×25 mL)萃取,合并有机层,无水硫酸钠干燥,减压除去溶剂,经柱层析(石油醚-乙酸乙酯,4∶1~1∶1)得到化合物22~28,其收率、理化常数及波谱数据见表1~2。
在目标化合物4~10的合成中,该反应是经过两步反应进行,如图4所示,首先苯甲氰化合物与羟胺反应生成胺肟中间体I,接着在微波的环境下与化合物3反应制得嘧啶类化合物4~10。目标化合物18是通过氧化相应的二氢吡唑类化合物17制备的;在合成咪唑类化合物22~28时,其中关键中间体21是通过采用POCl3-DMF试剂经过Vilsmeier-Haack反应制得的。
表1 目标化合物4~18,22~28的理化常数和质谱数据
Tab. 1 Physical contants and MS of synthesized compounds 4-18, 22-28

表2 目标化合物4~18,22~28的核磁氢谱数
Tab. 2 The1H-NMR data of target compounds 4-18, 22-28

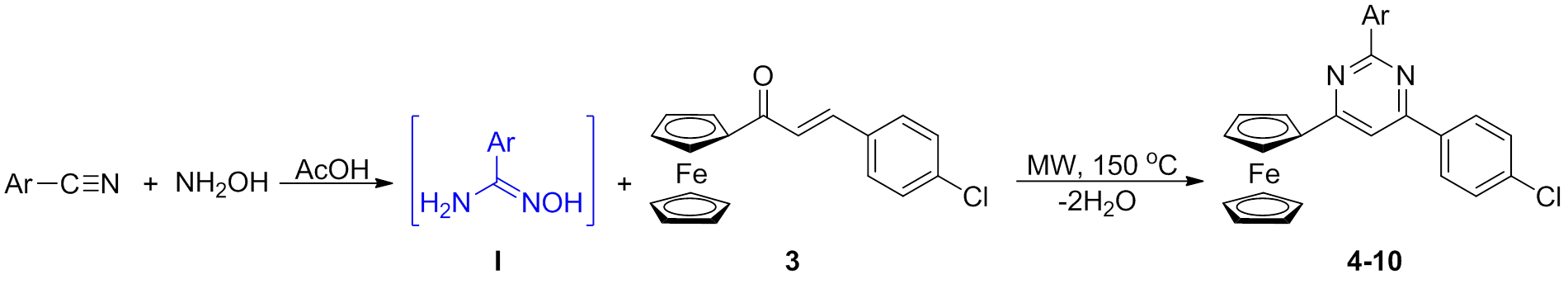
图4 二茂铁类化合物4~10的合成过程
Fig. 4 Synthetic route of ferrocenyl compounds 4-10
所合成的二茂铁类化合物均经1H-NMR和MS分析确证。在化合物4~10的核磁共振氢谱中,在7.9 ppm附近出现一个单峰归属于嘧啶环上氢的特征信号;在化合物11~18氢谱中,其二氢吡唑环上的三个烷基氢信号分别在5.5,3.8和3.0 ppm附近出现,而用DDQ将二氢吡唑环氧化成吡唑环后,3个烷基氢信号消失,并在6.8 ppm附近出现一个芳香质子的信号,归属于吡唑环上的氢质子信号;在化合物22~38氢谱中,出现一对反式双键的信号(J>15.0 Hz),归属于化合物中a,b-不饱和酮的双键质子特征信号。通过MS分析发现化合物测得的分子量与理论值一致,这说明所合成的化合物是目标产物。
选取MCF-7、MDA-MB-231这2种人乳腺癌细胞株和正常乳腺上皮细胞MCF-10A为测试细胞株,以他莫昔芬为阳性对照药,采用CCK8试剂盒测试合成的二茂铁衍生物抗乳腺癌活性。取对数生长期的MCF-7或MDA-MB-231细胞悬浮于含10%胎牛血清的无酚红DMEM培养基中,铺至96孔细胞培养板中。待细胞完全贴壁后,弃去原培养液,加入待测查耳酮,其浓度梯度为1×10-9,5×10-8,1×10-8,5×10-7,1×10-7,5×10-6,1×10-6,5×10-5,1× 10-5,1×10-4mol·L-1。药物处理培养3 d后,每孔加入5 μL CCK8,置于37 ℃、5% CO2培养箱中继续孵育4 h。使用酶标仪在490 nm波长测定每孔的吸光度(OD)值,分析实验结果,并计算出IC50,结果见表3。同时,为了考察目标化合物的毒性,测试了目标化合物对正常乳腺上皮细胞MCF-10A的抑制作用,并计算出这些化合物的治疗指数(in vitrotherapeutic index,IVTI)。化合物的IVTI值越高,安全性越大[12],结果见表3。
表3 目标化合物4~18,22~28对2种乳腺癌和MCF-10A细胞株的增殖抑制活性(n=3)
Tab. 3 Antiproliferative activities of target compounds 4-18, 22-28 on two breast cancer and MCF-10A cell lines (n=3)
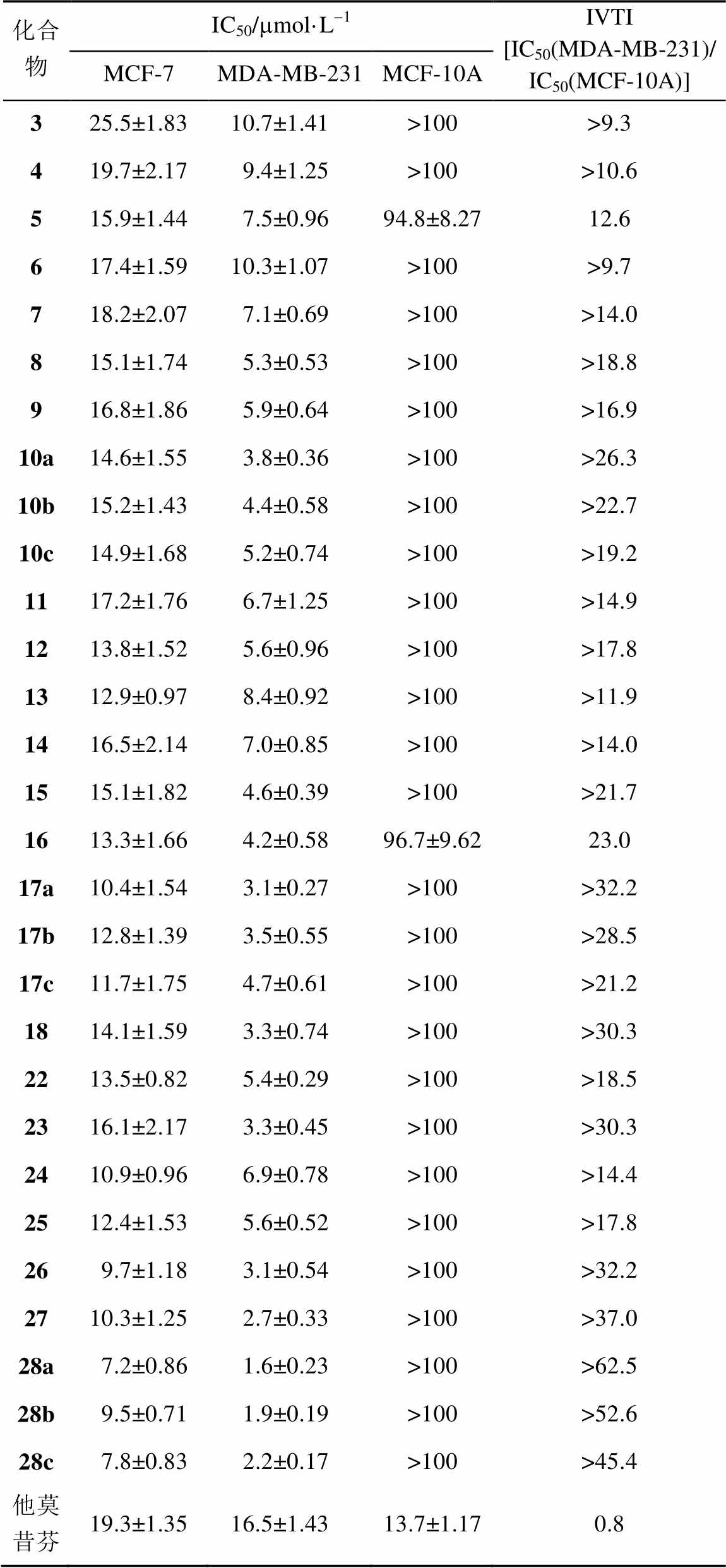
从上述活性结果可以发现:①目标二茂铁衍生物对荷尔蒙依赖型乳腺癌MCF-7细胞和TNBCMDA-MB-231细胞均有较强抑制活性,并显著强于先导化合物3。同时,这些二茂铁衍生物对MDA-MB-231细胞展现出了更强的选择性和抑制活性,其抑制MDA-MB-231的活性(IC50< 11mmol·L-1)均显著地强于阳性药物他莫昔芬 (IC50=16.5mmol·L-1);②芳环(Ar)上的取代基对活性影响较大,总的来说,芳环含有吸电子基取代的化合物抗肿瘤活性强于相应的给电子基化合物,尤其是4-F取代的化合物28a[IC50=(1.6± 0.23)mmol·L-1]对MDA-MB-231展现出了最强的抑制活性,甚至是阳性药物他莫昔芬的10倍,而进一步将化合物28a芳环上的F从对位转移到间位[化合物28b,IC50=(1.9±0.19)mmol·L-1]或邻位[化合物28c,IC50=(2.2±0.17)mmol·L-1]均降低了抗癌活性,这些结果说明芳环上的对取代能够提高抗肿瘤活性。事实上,在嘧啶类化合物和二氢吡咯类化合物中,也有着同样的规律,即4-F取代化合物的抗肿瘤活性强于其相应的间位和邻位取代衍生物;③除了取代基外,化合物中的杂环对抗肿瘤活性也有较大的影响,其中咪唑杂环类化合物抗肿瘤活性强于相应的二氢吡咯、吡咯和嘧啶杂环化合物;④所合成的二茂铁衍生物对正常的乳腺上皮细胞没有毒性并有较高的治疗指数(IVTI>9),而他莫昔芬[IC50=(13.7±1.17)mmol·L-1]却展现出了较强的细胞毒性,其IVTI值仅为0.8。
本实验以前期获得的二茂铁查耳酮为先导化合物,对其进行结构修饰,合成了一系列具有不同杂环的二茂铁衍生物,这些目标化合物抗乳腺癌活性均强于其先导化合物,并对TNBCMBA- MD-231细胞展现出了较强的选择性和抑制活性,其中化合物28a对MBA-MD-231展现出了最强的活性,分别是先导化合物和他莫昔芬的6和10倍;此外,这些二茂铁衍生物对正常的乳腺上皮细胞MCF-10A没有毒性。这些结果说明,对查耳酮的a,b-不饱和酮进行修饰能够提高其抗肿瘤活性,这为今后发展新型、高效、低毒的抗TNBC药物提供了新的思路。
REFERENCES
[1] Burnett J P, LIM G, LI Y, et al. Sulforaphane enhances the anticancer activity of taxanes against triple negative breast cancer by killing cancer stem cells [J]. Cancer Lett, 2017(394): 52-64.
[2] COSTA R, SHAH A N, SANTA-MARIA C A, et al. Targeting epidermal growth factor receptor in triple negative breast cancer: New discoveries and practical insights for drug development [J]. Cancer, 2017(53): 111-119.
[3] XU Y, GAO X. Influence of the medication administration sequence of docetaxel and epirubicin on the therapeutic effect during chemotherapy for breast cancer [J]. Chin J Mod Appl Pharm(中国现代应用药学), 2015, 33(6): 745-747.
[4] CHI Q Q, GENG W Q, ZHANG H, et al. Preparation and characteristics of glaucocalyxin A-loaded nanoliposomes and its inhibition of triple negative breast cancer(TNBC) [J]. Chin J New Drugs(中国新药杂志), 2016, 25(23): 2724-2731.
[5] WANG Y CHAN F L, CHEN S, et al. The plant polyphenol butein inhibits testosterone-induced proliferation in breast cancer cells expressing aromatase [J]. Life Sci, 2005, 77(1): 39-51.
[6] TAN J. Synthesis of chalcone derivatives and anti-breast cancer screening [J]. Chin J Mod Appl Pharm(中国现代应用药学), 2016, 33(3): 318-325.
[7] CHEN S L, TAN J. Synthesis of ferrocenyl chalcone compounds and their anti-breast cancer activities [J]. Drugs Clin(现代药物与临床), 2015, 30(8): 913-916.
[8] KAPLANCIKI Z A, TURAN-ZITOUNI G, OZDEMIR A, et al. Synthesis and antinociceptive activities of some pyrazoline derivatives [J]. Eur J Med Chem, 2009, 44(6): 2606-2610.
[9] LEBLANC R, DICKSON J, BROWN T, et al. Synthesis and cytotoxicity of epoxide and pyrazole analogs of the combretastatins [J]. Bioorg Med Chem, 2005, 13(21): 6025-6034.
[10] KERR D J, HAMEL E, JUNG M K, et al. The concise synthesis of chalcone, indanone and indenone analogues of combretastatin A4 [J]. Bioorg Med Chem Lett, 2007, 15(9): 3290-3298.
[11] LEE L, DAVIS R, VANDERHAM J, et al. 1,2,3,4- Tetrahydro-2-thioxopyrimidine analogs of combretastatin-A4 [J]. Eur J Med Chem, 2008, 43(9): 2011-2015.
[12] MULLER P K, MOLTON M N. The determination and interpretation of the therapeutic index in drug development [J]. Nat Rev Drug Discov, 2012, 11(10): 751-761.
(本文责编:蔡珊珊)
Synthesis of Ferrocenyl Heterocyclic Derivatives and Anti-triple Negative Breast Cancer Screening
TAN Juan, CHEN Ling, PENG Anlin, XIAO Jing, ZHANG Enjing
(Department of Medicine, Wuhan Third Hospital, TongRen Hospital of WuhanUniversity, Wuhan 430060, china)
ABSTRACT: OBJECTIVETo design and synthesis of ferrocenyl heterocyclic derivatives and investigate anti-triple negative breast cancer activity.METHODSA series of ferrocenyl derivatives were designed and synthesized from ferrocenyl chalcone, and their anti-breast cancer activities were evaluated by CCK8 assay.RESULTSTwenty-eight ferrocenyl heterocyclic derivatives were synthesized and the structures had been confirmed by1H-NMR and MS spectra. The preliminary biological results showed that all synthesized ferrocenyl derivatives showed selective anticancer activity that were more potent against MDA-MB-231 cells than MCF-7, which also showed moderate inhibitory activity, against MDA-MB-231 cell lines, and imidazole heterocyclic compounds had more potent anti-tumor than corresponding pyrazole and pyrimidine derivatives, specifically, compound 28a [IC50=(1.6±0.23)μmol·L-1] showed about 6 and 10-fold potency than lead compound 3 [IC50=(10.7± 1.41)μmol·L-1] and tamoxifen [IC50=(13.7±1.17)μmol·L-1], against MDA-MB-231 cell lines, and these ferrocenyl derivatives were not toxic to normal cells.CONCLUSIONThis study provides information and basis for development of ferrocenyl derivatives with anti-triple negative breast cancer activity.
KEY WORDS:ferrocenyl derivatives; synthesis; anti-triple negative breast cancer
中图分类号:R914.2
文献标志码:B
文章编号:1007-7693(2018)03-0345-07
DOI:10.13748/j.cnki.issn1007-7693.2018.03.009
收稿日期:2017-04-27
引用本文:谭娟, 陈灵, 彭安林, 等. 二茂铁杂环类化合物的合成及抗三阴性乳腺癌活性研究[J]. 中国现代应用药学, 2018, 35(3): 315-351.
作者简介:谭娟,女,硕士生 Tel: (027)65399485 E-mail: juantan@126.com