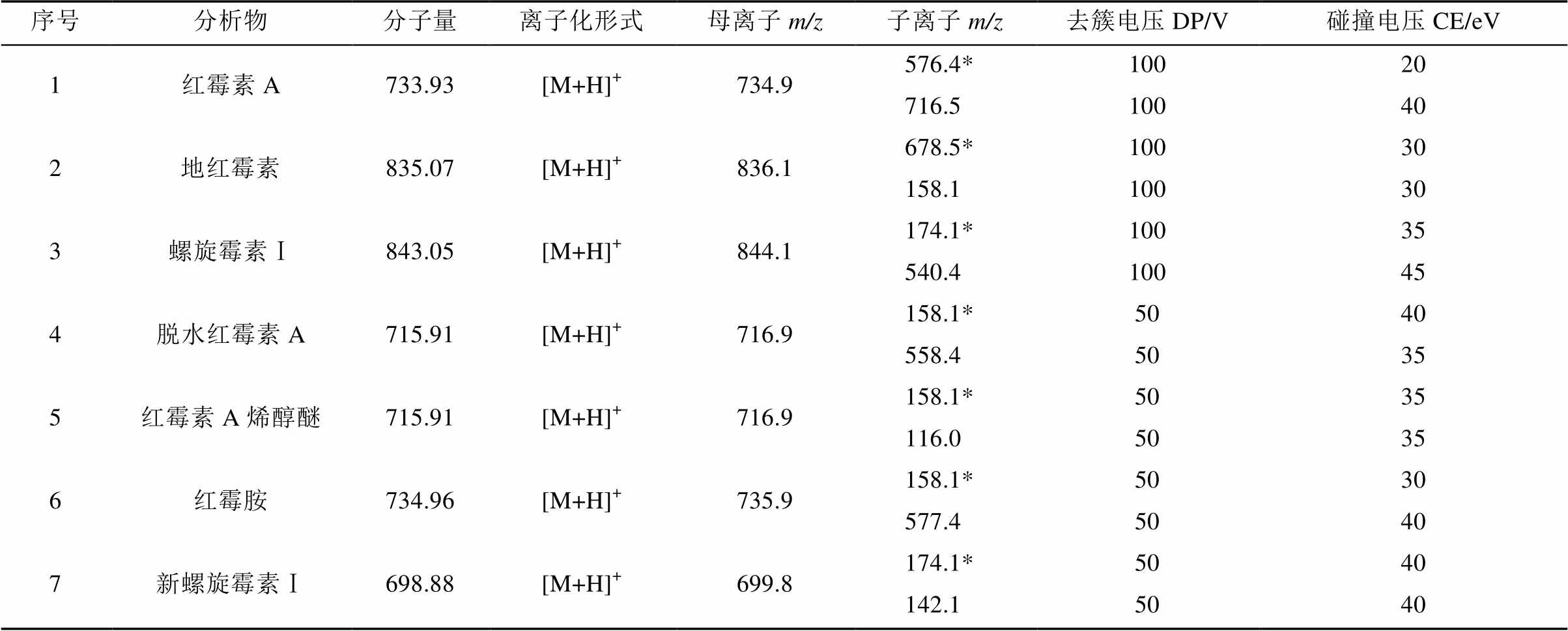
表1 3种大环内酯类抗菌药物及其代谢物的质谱条件
Tab. 1 The mass spetrum conditions of three macrolide antibiotics and their metabolites
王旭1,刘国婷2,宋金芝3
(1.天津医科大学第二医院药学部,天津 300210;2.天津市北辰医院药学部,天津 300400;3.天津西青医院药学部,天津 300380)
摘要:目的 建立测定人血浆中3种大环内酯类抗菌药物(红霉素、地红霉素、螺旋霉素Ⅰ)及其代谢物(脱水红霉素、红霉素A烯醇醚、红霉胺、新螺旋霉素Ⅰ)的分析方法。方法 用乙腈溶液提取,QuECHERS方法净化,采用无水硫酸镁和无水乙酸钠沉淀蛋白和盐析分层,无水硫酸镁和石墨碳烯净化,上清液氮气吹干后甲醇复溶待测,用Eclipse Plus C18色谱柱分离,电喷雾正离子模式,多反应离子监测扫描,内标法定量。结果 3种大环内酯类抗菌药物及其代谢物浓度在4~ 900 ng·mL-1内的线性关系良好(r>0.999),定量限<10 ng·mL-1;在3个添加水平下,3种大环内酯类抗菌药物及其代谢物的提取回收率可达81.45%~96.82%,日内精密度<5.00%,日间精密度<10.00%。结论 QuECHERS-液相色谱串联三重四级杆质谱测定血浆中3种大环内酯类抗菌药物及其代谢物的分析方法简单,快速,灵敏,特异性强,适用于血浆中大环内酯类抗菌药物及其代谢物的测定。
关键词:大环内酯类抗菌药物;代谢物;红霉素;地红霉素;螺旋霉素Ⅰ;高效液相色谱串联质谱;血浆
大环内酯类是一类具有14~16元大环内酯结构的抗菌类药物,主要有红霉素、螺旋霉素、地红霉素等[1-2]。其抗菌谱主要是革兰阳性菌、厌氧菌和部分革兰阴性菌及非典型病原体等,临床上主要用于治疗泌尿生殖系统等感染性疾病,支气管肺炎等呼吸系统疾病和胃肠动力障碍等消化系统疾病[3-5]。由于该类药物口服吸收好,体内分布广,组织浓度高,半衰期长,不良反应少[5-6],因而获得了广泛的临床应用。有研究报道红霉素与螺旋霉素共同治疗眼疾患者的金黄色葡萄球菌感染,两者具有协同作用[7],而地红霉素作为新一代大环内酯的长效药,常用于治疗急性化脓性扁桃体炎[8]。当患者同时服用红霉素、螺旋霉素和地红霉素时,三者药动学特点尚不明确,不利于临床应用。为了进一步指导临床应用,需要监测其血药浓度。目前测定大环内酯类抗菌药物的分析方法主要是应用HPLC和HPLC-MS法[9-14],而大环内酯类抗菌药物的代谢物在血浆中的含量较低,需要高灵敏度的仪器才能检测,因此需要应用HPLC-MS测定微量或痕量的大环内酯类抗菌药物及其代谢物。目前,红霉素、螺旋霉素和地红霉素3种药物在临床上应用较广泛,他们的代谢物保持了原药相似的结构,具有一定的药理活性,其代谢物不容忽视[15-17]。大多数文献报道了测定大环内酯类抗菌药物原药的分析方法,但尚未有相关文献报道同时测定3种大环内酯类抗菌药物及其代谢物含量的分析方法。为了更加全面检测血浆中大环内酯类抗菌药物的代谢情况并反映临床疗效,原药与代谢物含量有必要同时监测。Quick, Easy, Cheap, Effective, Rugged, and Safe extraction (QuECHERS)样品前处理由Anatassiades等[18]于2003年提出,采用MgSO4盐析分层和分散固相萃取净化样品,最初主要用于农产品里农药残留分析中,随后应用于食品领域净化奶制品中药物残留分析,该法具有简单、快速、经济等优势而逐渐被应用于样品净化。目前应用QuECHERS方法处理血浆样品的大环内酯类抗菌药物的研究尚未有报道,因此本研究建立HPLC-MS测定人血浆中3种大环内酯类抗菌药物及其代谢物含量分析方法,为药动学研究和临床用药进一步提供依据。
Agilent 1260型高效液相色谱系统,包括二元泵,在线脱气,自动进样器(美国Agilent公司);Applied Biosystems Qtrap 5500三重四级杆质谱仪(加拿大AB公司);BS224S型电子天平(北京赛多利斯仪器系统有限公司);KQ-500DE型超声仪(昆山市超声仪有限公司);TG16-WS离心机(长沙市平凡仪器有限公司);N-EVAP116型氮气浓缩仪(美国Organomation Associates公司);Agilent Eclipse Plus-C18色谱柱(2.1 mm×150 mm,3.5 μm,美国Agilent 公司);0.22 µm尼龙滤膜(天津津腾有限公司)。
红霉素对照品(德国Dr. Ehrenstorfer GmbH,批号:40527,纯度:99.5%);螺旋霉素对照品(德国Dr. Ehrenstorfer GmbH,批号:31022,纯度:99.0%);地红霉素对照品(中国食品药品检定所,批号:PX64-MONM,纯度:99.6%);新螺旋霉素对照品(批号:8-QFY-26-1,纯度>99%)、红霉胺对照品(批号:2-OMK-3-1,纯度>99%)、脱水红霉素A对照品(批号:1-ALB-97-1,纯度>99%)、红霉素A烯醇醚对照品(批号:3-YEN-17-2,纯度>99%)均购自加拿大TRC公司;罗红霉素(德国Dr. Ehrenstorfer公司,批号:30824);甲醇和乙腈(色谱纯,美国Thermo Fisher公司);甲酸、甲酸铵(色谱纯,德国Sigma-Aldrich公司);乙二胺-N-丙基硅烷(Primary-secondary amine,PSA,德国Sigma-Aldrich公司);C18固体粉末(德国CNW Technologies GmbH公司);石墨碳烯(PestiCarb,PC);固体粉末(美国Agilent 公司)。
取天津医科大学第二医院健康体检者血液,加入抗凝剂肝素,抗凝剂∶血液=1∶9,混匀,离心(离心半径为6 cm,离心5 min,转速为5 000 r·min-1)后所得的上清液即为血浆。
2.1 液相色谱串联质谱条件
2.1.1 色谱条件 色谱柱:Agilent Eclipse Plus-C18(2.1 mm×150 mm,3.5 μm);柱温:室温;进样量:5 μL;流速:0.5 μL·min-1;流动相:0.1%甲酸水溶液(A)-乙腈(B)。梯度洗脱,洗脱程序:0~1 min,90%→85% A;1~15 min,85%→55% A;15~16 min,55%→90% A;16~20 min,90% A。
2.1.2 质谱条件 离子源类型:点喷雾电离源ESI(+);电喷雾电压:5 000 V;离子源温度:500 ℃;雾化气压力:50 kPa;辅助气压力:55 kPa;气帘气压力:32 kPa;碰撞室入口电压:10 V;碰撞室出口电压:7 V;碰撞电压、去簇电压、定性离子对、定量离子对、化合物分子量见表1。
2.2 贮备液对照品溶液的制备
分别准确称取大环内酯类抗菌药物(红霉素、地红霉素、螺旋霉素Ⅰ)及其代谢物(脱水红霉素、红霉素A烯醇醚、红霉胺和新螺旋霉素Ⅰ)(2.5± 0.2) mg,分别置于25 mL量瓶中,用甲醇稀释并定容,得0.1 mg·mL-1的大环内酯类抗菌药物及其代谢物的贮备液,另外配置内标罗红霉素标准贮备液(2.5±0.2)mg置于25 mL量瓶中,得0.1 mg·mL-1内标贮备液,均置于-20 ℃冰箱中避光冷冻保存待用。
表1 3种大环内酯类抗菌药物及其代谢物的质谱条件
Tab. 1 The mass spetrum conditions of three macrolide antibiotics and their metabolites
注:*-定量离子。
Note: *-quantitative ion.
2.3 混合对照品工作液的制备
分别精密移取500 µL红霉素A、700 µL地红霉素、600 µL螺旋霉素Ⅰ、500 µL脱水红霉素A、400 µL红霉素A烯醇醚、700 µL红霉胺、900 µL新螺旋霉素Ⅰ的“2.2”项下贮备液和内标1 000 µL,分别至10 mL量瓶中,用甲醇溶解并定容,混匀后即得红霉素A 5 µg·mL-1、地红霉素7 µg·mL-1、螺旋霉素Ⅰ6 µg·mL-1、脱水红霉素A 5 µg·mL-1、红霉素A烯醇醚4 µg·mL-1、红霉胺7 µg·mL-1、新螺旋霉素Ⅰ9 µg·mL-1大环内酯类抗菌药物混合对照品溶液,以及10 µg·mL-1的罗红霉素内标溶液,于4 ℃冰箱中保存待用。分别精密移取10,20,50,100,200,500,1 000 µL混合对照品溶液至10 mL量瓶中,并将100 µL的10 µg·mL-1的罗红霉素内标溶液加入每个浓度混合对照品溶液中,用甲醇定容至10 mL,可得红霉素A 5,10,25,50,100,250,500 ng·mL-1,地红霉素7,14,35,70,140,350,700 ng·mL-1,螺旋霉素Ⅰ6,12,30,60,120,300,600 ng·mL-1,脱水红霉素A5,10,25,50,100,250,500 ng·mL-1,红霉素A烯醇醚4,8,20,40,80,200,400 ng·mL-1,红霉胺7,14,35,70,140,350,700 ng·mL-1,新螺旋霉素Ⅰ9,18,45,90,180,450,900 ng·mL-1,并含内标1 000 ng·mL-1的对照品线性工作液,用于当天实验使用。
2.4 供试品溶液
采用改良的QuEChERS方法进行前处理。准确移取人血浆200 µL置于50 mL离心瓶中,加入乙腈10 mL,涡旋1 min,分别加入硫酸镁500 mg和无水硫酸钠400 mg用于沉淀蛋白和盐析作用,涡旋1 min,混匀,离心半径为6 cm,离心(r=5 000 r·min-1) 5 min,提取上清液10 mL至第2根50 mL离心管中,然后将无水硫酸镁500 mg和石墨碳烯40 mg分别加入第2个离心管中,涡旋1 min。然后吸取5 mL上清液至10 mL试管,在40 ℃水浴锅中氮吹至干,用500 µL甲醇溶解残留物,用0.22 µm尼龙滤膜过滤后上机检测。
2.5 方法学考察
2.5.1 方法专属性考察 分别取5份空白血浆200 μL,按“2.4”项下供试品溶液的操作步骤处理,根据“2.1.1”项和“2.1.2”项下色谱和质谱条件,进样5 μL测量。结果表明,血浆内源性成分不干扰3种大环内酯类抗菌药物及其代谢物和内标测定,同时各待测成分分离良好,该前处理方法和HPLC-MS适宜血浆中大环内酯类抗菌药物及其代谢物的定性和定量。色谱图见图1。
2.5.2 线性关系和定量限 在“2.1.1”项和“2.1.2”项下色谱和质谱条件下,用空白血浆加入对照品溶液配制各大环内酯类抗菌药物及其代谢物的血浆样品线性范围溶液,按“2.4”项下方法处理样品,建立3种大环内酯类及其代谢物标准曲线。以提取离子色谱峰的峰面积(Y)作为纵坐标,质量浓度(X)为横坐标建立标准线性方程,并以内标法定量。将各大环内酯类抗菌药物及其代谢物标准溶液逐步稀释至所测的信号为10倍的噪音时,可得测其浓度为定量限。结果表明,3种大环内酯类抗菌药物及其代谢物在线性范围内线性良好,r>0.999,定量限<10 ng·mL-1,说明该方法适合3种大环内酯类抗菌药物及其代谢物的定量分析。结果见表2。
2.5.3 日内、日间精密度 取空白血浆配制3种大环内内酯类抗菌药物及其代谢物线性范围的第2,4,6个浓度的3个添加水平的血浆样品溶液,每个浓度的样品溶液平行制备6份,测定结果计算作为日内精密度。根据当天的标准曲线,连续测3 d,测定结果作为日间精密度。结果表明日内精密度<5.00%,日间精密度均<10.00%。结果见表3。
2.5.4 提取回收率和基质效应 对照品溶液直接上机测定,测得的峰面积为A。取6份空白血浆200 µL,根据“2.4”项下供试品前处理操作到溶解残渣前,其残渣加入适量标准溶液配制成各大环内酯类抗菌药物及其代谢物线性范围第2,4,6个浓度的3个添加水平的样品溶液(内含100 ng·mL-1内标溶液),用甲醇定容后上机测定,测得的峰面积为B。取6份空白血浆200 µL,加入适量标准溶液配制成各大环内酯类抗菌药物及其代谢物线性范围第2,4,6个浓度的3个添加水平的样品溶液,根据“2.4”供试品前处理后的样品溶液上机测定,测得的峰面积为C。基质效应=B/A×100%;提取回收率为C/B×100%。3种大环内酯类抗菌药物及其代谢物的基质效应在81.02%~115.66%;内标的提取回收率为81.45%~96.82%。这表明该色谱和质谱条件下,基质效应的影响可以忽略。提取回收率均符合生物样品分析的要求。结果见表4。
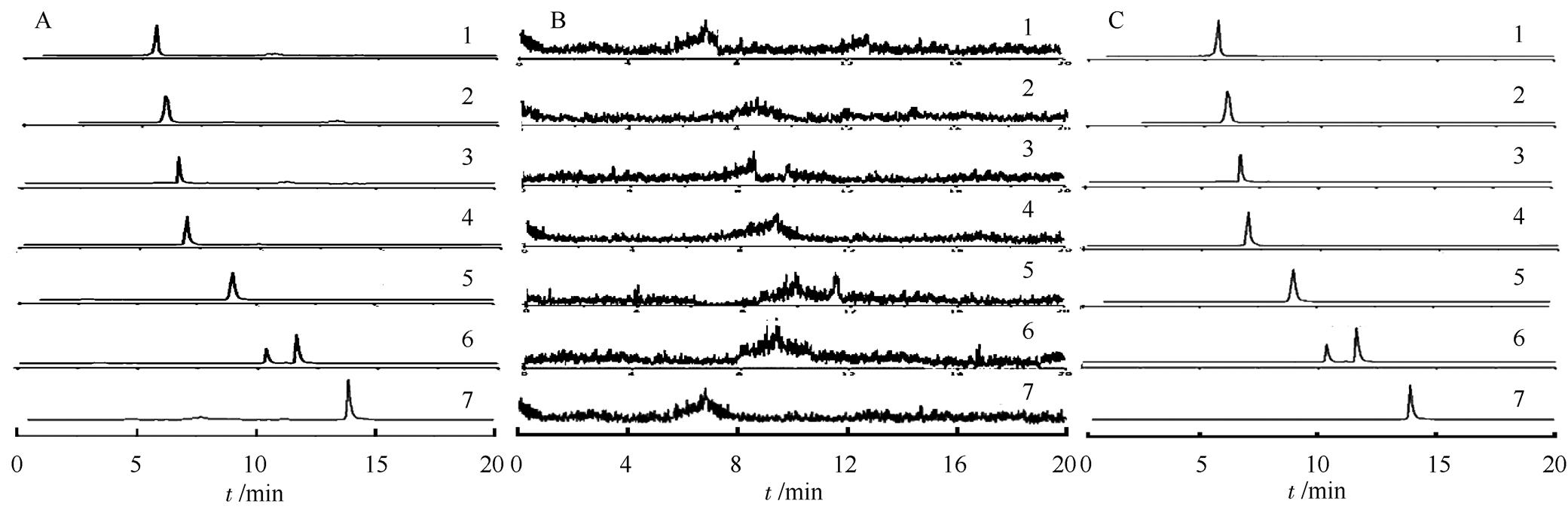
图1 3种大环内酯类抗菌药物及其代谢物的提取离子流图
A-空白血浆+3大环内酯类抗菌药物及其代谢物对照品;B-空白血浆;C-3大环内酯类抗菌药物及其代谢物对照品;1-新螺旋霉素;2-螺旋霉素;3-红霉胺;4-地红霉素;5-红霉素A;6-脱水红霉素和红霉素A烯醇醚;7-罗红霉素。
Fig. 1 The chromatograms of extracted ion current for three macrolide antibiotics and their metabolites
A blank plasma+reference substances of three marolide antibiotics and their metabolites; B-blank plasma; C-reference substances of three marolide antibiotics and their metabolites; 1-neospiramycin; 2-spiramycin; 3-erythromycylamine; 4-dirithromycin; 5-erythromycin A; 6-anhydroerythromycin A and erythromycin A ene alcohol ether; 7-roxithromycin.
表2 3种大环内酯类抗菌药物及其代谢物的线性方程
Tab. 2 The regression equation, the limit of detection and the limit of quantification for three macrolide antibiotics and their metabolites

表3 3种大环内酯类抗菌药物及其代谢物的日内、日间精密度
Tab. 3 The results of precision for three macrolides and their metabolites
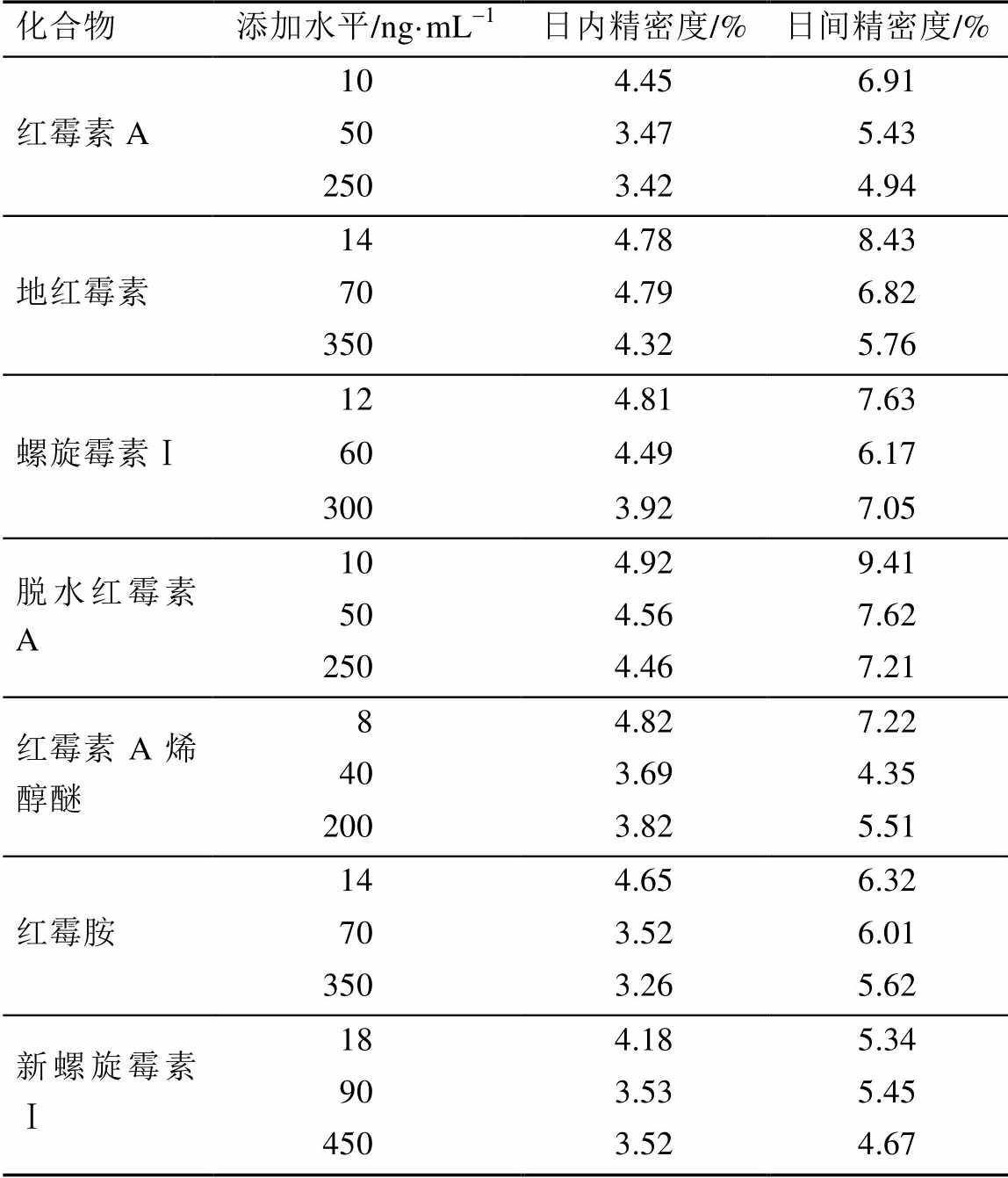
表4 3种大环内酯类抗菌药物及其代谢物的基质效应和提取回收率
Tab. 4 The reaults of matrix effects and extraction recoveries for three macrolide antibiotics and their metabolites

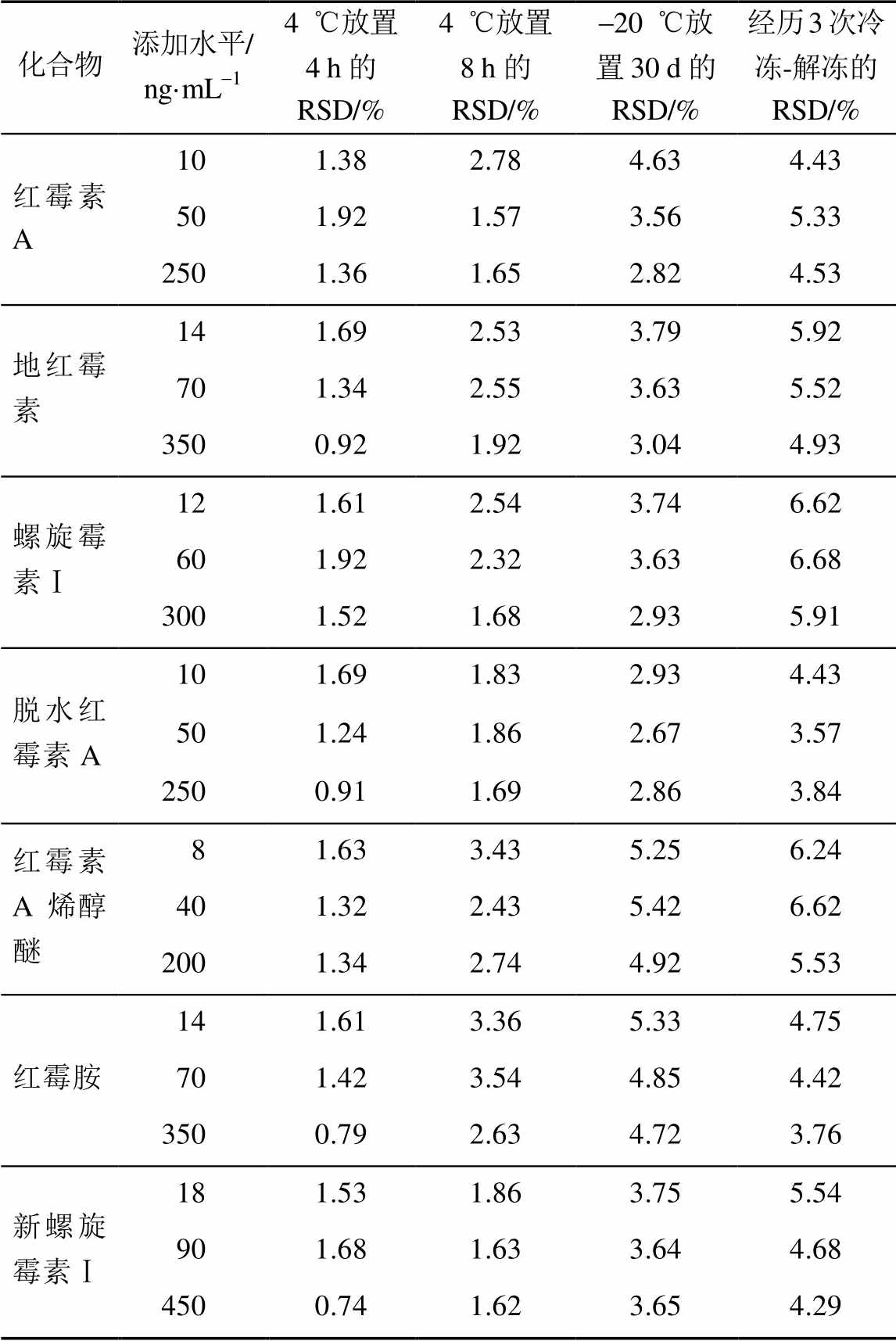
2.5.5 稳定性试验 本实验考察了各大环内酯类抗菌药物及其代谢物线性范围第2,4,6个浓度的3个添加水平的样品溶液在不同保存条件下的稳定性(n=3)。结果表明,血浆样品经过处理后4 ℃放置4 h的RSD均<7.00%,稳定;血浆样品经过处理后4 ℃放置8 h的RSD均<7.00%,稳定;血浆样品经过处理后在-20 ℃放置30 d的RSD均<7.00%,稳定。样品经历3次冷冻-解冻循环的RSD均<7.00%,稳定。结果见表5。
表5 3种大环内酯类抗菌药物及其代谢物的稳定性
Tab. 5 The results of stability for three macrolides and their metabolites
3.1 质谱条件的优化
该实验分别优化了负离子模式和正离子模式,结果表明,正离子模式的响应更高,更有利于质子化。这可能是因为大环内酯类抗菌药物大多数是个大环内酯环通过羟基与去甲氨基糖缩合生成的碱性苷,其中大环内酯环一般含有羟基、酮基或醛基等,这些基团在ESI离子源的正离子模式下具有较大的响应[19-20]。螺旋霉素、新螺旋霉素的化学结构中含有2个氮原子。在Q1扫描过程中能够生成[M+H]+和[M+2H]2+,而[M+H]+形式响应比较大。在子离子扫描中,通过离子响应强度大小,选择3~4个较大响应的子离子,供MRM扫描时选择,最后选择响应较大的2个子离子作定性离子,并从中选择一个响应较大的离子作为定量离子。此外脱水红霉素A和红霉素A烯醇醚是同分异构体,不仅具有相同的分子式,在质谱中也能产生3个相同的子离子,分别是m/z158.1,m/z116.0,m/z558.4。通过液相色谱条件的优化,红霉素的代谢物脱水红霉素A和红霉素A烯醇醚能够实现良好分离而分别定性和定量,他们的保留时间分别为10.58 min和13.65 min。
3.2 液相色谱条件的优化
实验分别考察了乙腈和甲醇流动相,乙腈在Eclipse Plus-C18色谱柱下洗脱大环内酯类抗菌药物的能力比甲醇强。为了减少有机溶剂的使用和节省分析时间,最终选择乙腈-0.1%甲酸水溶液作为流动相,在20 min内3种大环内酯类抗菌药物及其代谢物能够实现良好的分离,特别是脱水红霉素和红霉素A烯醇醚能够实现良好的分离,不受杂质的影响,大多数分离度均能>1.5,如图1所示,符合检测的需要。
3.3 样品前处理条件的优化
根据大环内酯类抗菌药物易溶于极性溶剂的性质,本实验考察乙腈、甲醇、0.1%甲酸乙腈溶液、和0.1%甲酸-5 mmol·L-1甲酸铵乙腈溶液提取大环内酯类抗菌药物及其代谢物。结果表明,用甲醇提取血浆中的多数大环内酯类抗菌药物回收率<40%。而0.1%甲酸乙腈溶液、5 mmol·L-1甲酸铵乙腈或0.1%甲酸-5 mmol·L-1甲酸铵乙腈溶液提取血浆中的大环内酯类抗菌药物时,红霉素及其代谢物等一些大环内酯类抗菌药物容易受酸碱的影响,造成回收率不稳定或者回收率不理想。用乙腈为提取溶剂时,3大环内酯类抗菌药物的回收率可>80%,同时乙腈也可以达到沉淀蛋白的作用。因此最终选择乙腈提取血浆中的大环内酯类抗菌药物。实验还分别考察了5,10,15,20,25,30和35 mL乙腈溶液的提取效率。结果显示,10~15 mL乙腈整体提取效率较合理,为了节省溶剂,最终选择10 mL。但是单纯用乙腈提取,基质效应较大,不利于定量,特别是地红霉素、红霉胺、螺旋霉素和新螺旋霉素,四者的基质效应分别为34.89%,58.38%,139.37%和127.17%。因此需要进行进一步的净化。由于QuEChERS前处理方法具有操作简便、快速、经济等优势,因此本实验采用该法。本实验分别考察传统的QuEChERS方法和改良后QuEChERS方法。在第2个净化步骤中,改良后QuEChERS方法是考察石墨碳烯(PestiCarb,PC)材料,而传统的QuEChERS方法是采用乙二胺-N-丙基硅烷(Primary–secondary amine,PSA)和C18材料,分别考察3者对基质效应和提取回收率的影响。结果表明,PC与C18的除杂效果与相当,二者均比PSA除杂效果好。同时从实验结果得知,对于大多数大环内酯类抗菌药物,PC比单一C18材料、单一PSA材料、和C18+PSA组合材料的基质效应和提取回收率更合理,因此选择PC净化样品,基质效应和提取回收率均符合检测分析需要。
3.4 临床应用
该方法的建立为测定人血浆中3种大环内酯类抗菌药物及其代谢物含量提供检测方法,为应用于临床实验的药动学研究提供基础,但是还未实际应用临床试验中,有待更深入的研究。
[1] Song X, Zhou T, Liu Q. Molecularly imprinted solid-phase extraction for the determination of ten macrolide drugs residues in animal muscles by liquid chromatography-tandem mass spectrometry [J]. Food Chem, 2016(208): 169-176.
[2] SUN S J. The progress of clinical application of macrolides [J]. Chin Lic Pharm(中国执业药师), 2011, 8(6): 19-21, 38.
[3] ZHOU Y, Zhou T, JIN H. Rapid and selective extraction of multiple macrolide antibiotics in foodstuff samples based on magnetic molecularly imprinted polymers [J]. Talanta, 2015(137): 1-10.
[4] HU Y. Clinical value of macrolide antibiotics in the treatment of children with bronchopneumonia [J]. J Clin Pulm Med(临床肺科杂志), 2013, 18(3): 496-497.
[5] LI Z Y, CUI Y B, ZHANG J X. Progress in studies of macrolide antibotics [J]. World Notes Antibiotics(国外医药抗生素分册), 2013, 34(1): 6-15.
[6] 葛涵, 沈舜义. 大环内酯类抗生素研究进展[J]. 世界临床药物, 2007, 28(6): 376-380.
[7] DENG X H, GUO Y W, SONG H Y. Effect of erythromycin and spiramycin on ocular staphylococcus aureus biofilm [J]. Rec Adv Ophthalmol(眼科新进展), 2012, 32(9): 834-836.
[8] 金晶, 袁玉林, 叶林峰, 等. 地红霉素联合尼美舒利治疗急性化脓性扁桃体炎42例[J]. 医药导报, 2010, 29(12): 1595-1596.
[9] G VINCENT U, VON HOLST C. Sample preparation strategy for the simultaneous determination of macrolide antibiotics in animal feedingstuffs by liquid chromatography with electrochemical detection (HPLC-ECD) [J]. J Pharm Biomed Anal, 2007, 43(5): 1628-1637.
[10] TORANO J S, GUCHELAAR H J. Quantitative determination of the macrolide antibiotics erythromycin, roxithromycin, azithromycin and clarithromycin in human plasma by high-performance liquid chromatography using pre-column derivatization with 9-fluorenylmethyloxycarbonyl chloride and fluorescence detection [J]. J Chromatography B: Biomed Sci Appl, 1998, 720(1): 89-97.
[11] 赵东豪, 贺利民, 聂建荣, 等. HPLC-MS/MS检测猪肉中六种大环内酯类抗生素[J]. 分析试验室, 2009, 28(1): 117-119.
[12] JANK L, MARTINS M T, ARSAND J B. High-throughput method for macrolides and lincosamides antibiotics residues analysis in milk and muscle using a simple liquid-liquid extraction technique and liquid chromatography-electrospray- tandem mass spectrometry analysis (LC-MS/MS) [J]. Talanta, 2015(144): 686-695.
[13] CAZORLA-REYES R, ROMERO-GONZALEZ R, FRENICH AG. Simultaneous analysis of antibiotics in biological samples by ultra high performance liquid chromatography- tandem mass spectrometry [J]. J Pharm Biomed Anal, 2014(89): 203-212.
[14] LI W, RETTING J, JIANG X. Liquid chromatographic- electrospray tandem mass spectrometric determination of clarithromycin in human plasma [J]. Biomed Chromatogr, 2006, 20(11): 1242-1251.
[15] 刘茜, 王桂凤, 陈天书, 等. 液相色谱-串联质谱法测定人血浆中红霉胺的含量 [J]. 沈阳药科大学学报, 2009, 26(5): 393-396.
[16] ZHANG X Y, ZHENG D Z, YUAN F. Determination of erythromycin A and its metabolites in honey by high performance liquid chromatography-tandem mass spectrometry [J]. J Instrum Anal(分析测试学报), 2013, 32(9): 1131-1134.
[17] WANG J, LEUNG D. Short communication determination of spiramycin and neospiramycin antibiotic residues in raw milk using LC/ESI-MS/MS and solid-phase extraction [J]. J Sep Sci, 2009(32): 681-688.
[18] ANASTASSIA M, LEHOTAY S J, STAJNBAHER D. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and "dispersive solid-phase extraction" for the determination of pesticide residues in produce [J]. J AOAC Int, 2003, 86(2): 412-31.
[19] LIU Y. HPLC analysis of macrolide antibiotic residues in animal derived food [D]. Jiangsu: Jiangnan University, 2008.
[20] WANG J. Analysis of macrolide antibiotics, using liquid chromatography-mass spectrometry, in food, biological and environmental matrices [J]. Mass Spec Rev, 2009, 28(1): 50-92.
(本文责编:蔡珊珊)
Wang Xu1, Liu Guoting2, Song Jinzhi3
(1.Pharmaceutical Department, The second hospital of tianjin medical university, Tianjin 300210, China; 2.Pharmaceutical department, Tianjin Beichen Hospital, Tianjin 300400, China; 3. Pharmaceutical department, Tianjin Xiqing Hospital, Tianjin 300380, China)
ABSTRACT:OBJECTIVE To establish a determination method of 3 macrolide antibiotics (erythromycin A, dirithromycin, spiramycin Ⅰ), and it’s metabolite (anhydroerythromycin A, erythromycin A ene alcohol ether, erythromycylamine, neospiramycin Ⅰ) in plasma using QuECHERS followed by HPLC-MS.METHODS The macrolide antibiotics in samples were extracted with acetonitrile. The plasma was directly purified by QuECHERS pretreatment method. MgSO4and sodium acetate anhydrous were added into plasma sample for precipitating protein and salting out stratification. Then, MgSO4and PestiCarb were used for dehydration and removing impurity. The upper layer was evaporated to dryness under a stream of nitrogen and the residue obtained was redissolved in methanol. three macrolide antibiotics were separated by Eclipse Plus C18chromatographic column. Electro spray ionization was used to analyze these macrolide antibiotics by using positive ion mode. Multiple reaction monitoring was used to monitor ions. Internal standard method was used to quantitative calculation. RESULTS three macrolide antibiotics showed good linear relationship in the range of 4-900 ng·mL-1(r>0.999). The quantitation limits was <10 ng·mL-1. At three levels, the average extract recoveries were 81.45%-96.82%, and the intra-day and inter-day precisions were respectively lower than 5.00% and 10.00%, respectively. CONCLUSION The analytical method of 3 macrolide antibiotics in plasma using QuEChERS and HPLC-MS is simple, rapid, sensitive and specific. The analytical method is suitable to quantitate macrolide antibiotics and their metabolites in plasma.
KEY WORDS:macrolide antibiotics; metabolites; erythromycin A; dirithromycin; spiramycin Ⅰ; HPLC-MS; plasma
中图分类号:R917.101
文献标志码:B
文章编号:1007-7693(2017)09-1289-07
DOI:10.13748/j.cnki.issn1007-7693.2017.09.016
引用本文:王旭, 刘国婷, 宋金芝. QuECHERS-HPLC-MS测定人血浆中3种大环内酯类抗菌药物及其代谢物含量[J]. 中国现代应用药学, 2017, 34(9): 1289-1295.
作者简介:王旭,女,硕士,药师 Tel: (022)88329098 E-mail: wangxu_sjz@163.com
收稿日期:2016-11-08