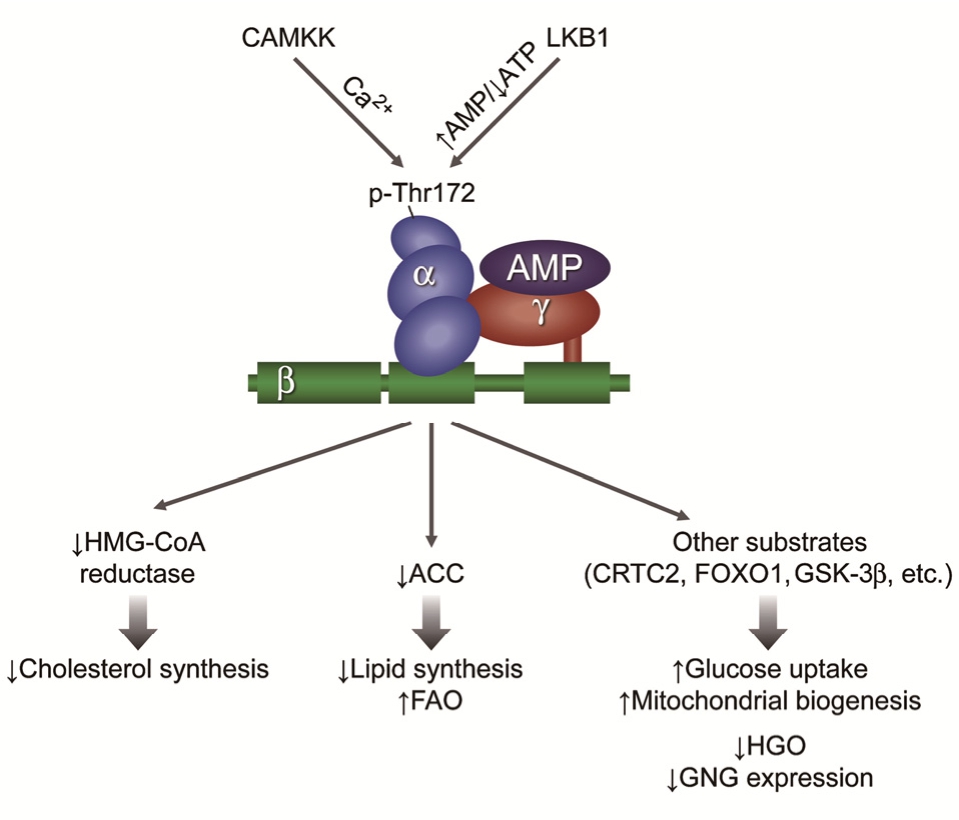
图1 AMPK的激活及调控[16]
Fig. 1 AMPK activation and regulation
陈金成1,2,张吟1*(1.福建医科大学附属第二医院药学部临床药学室,福建 泉州 362000;2.福建医科大学药学院,福州 350108)
摘要:腺苷酸活化蛋白激酶(AMP activated protein kinase,AMPK)是一种广泛参与多种代谢调节的激酶,是主要的细胞“能量感受器”。AMPK通过α亚基的Thr172发生磷酸化而激活,通过抑制脂质合成相关转录因子以及肝脏糖异生来控制脂质合成以及血糖稳定。肝脏糖异生的调控过程中,通过激素及转录因子如FoxO1、CREB、TORC2、HNF-4α、PGC-1α等的参与,最后将信号传达至糖异生的2个关键性限速酶基因PECK与G6Pase,起到调控作用。激活的AMPK能抑制糖异生转录因子及相关关键酶的有效表达,阻止肝脏发生糖异生,进而下降血糖浓度。本文旨在讨论与总结AMPK在肝脏糖脂代谢中的作用,重点分析AMPK与肝脏糖异生信号通路转录因子之间的联系。
关键词:腺苷酸活化蛋白激酶;肝脏糖脂代谢;糖异生;PEPCK;G6Pase;转录调节因子
近年来,糖尿病等代谢疾病的发病率不断增加,其主要发病原因与糖脂代谢密切相关。当前关于糖脂代谢紊乱的研究越来越多,涉及有西药及植物药等,如匡荣等[1]及董介正等[2]关于西药及植物药提取物对糖脂代谢紊乱的研究。近年来,深入研究与糖脂代谢有关的治疗靶点及信号通路的分子机制成为研究者关注的重点。研究表明,胰岛素抵抗是代谢类疾病主要的发病原因,而糖异生异常导致内源性葡萄糖输出增多又是其主要的病理生理特点[3],糖异生信号通路又受一系列转录因子调控。腺苷酸活化蛋白激酶(AMP activated protein kinase,AMPK)是一种广泛参与多种代谢调节的激酶,已成为研究糖尿病等代谢疾病的重要靶点[4]。研究证明:AMPK激活以后可以抑制糖异生,抑制内源性葡萄糖输出,降低空腹血糖[5],因此阐明AMPK与糖异生信号通路之间的联系对于研究糖尿病等代谢疾病的分子机制具有重要意义。
1.1 AMPK的结构
AMPK是一类异源三聚体的结构蛋白,广泛存在于真核细胞中,有α、β和γ亚基,其中α有α1和α2 2种亚基,β有β1和β2 2种亚基,γ有γ1、γ2和γ3 3种亚基。其α亚基是催化亚基,β和γ是调节亚基[6]。AMPK各亚基具有不同的分布部位,如α1一般可在哺乳动物的肝、肺、肾及心脏等处获得,而α2一般分布于动物的心脏、骨骼肌、肝脏中。研究发现,在糖脂代谢的调节中,AMPK的β和γ亚基不参与,α亚基在Thr172发生磷酸化后具有活性[7]。
在肝脏组织中,AMPK的α1和α2亚型都有表达,而α2起着重要的作用。研究发现,与α1对比,在α2基因敲除的小鼠中出现了高血糖及低胰岛素血症,可见AMPK的α2亚型在糖脂代谢过程中的重要作用[8]。
1.2 AMPK活性调节
1.2.1 AMPK上游激酶 AMPK是一种较为重要的蛋白激酶,且其具有较为复杂的活性调节能力,不仅能被磷酸肌酸别构抑制,也可被5'-AMP别构及其上游所存在的AMPK所激活[9]。至今,研究人员已发现至少3种AMPK上游激酶,如TGF-β活化激酶-1(TGF-b-activatedkinase-1,TAK1)、肿瘤抑制因子(liver kinase B1,LKB1)以及钙调蛋白依赖性蛋白激酶(Calmodulin- dependent protein kinasekinase,CaMKK)等。以上3种上游激酶均通过不同的方式在AMPK亚基上起作用[10]。LKB1可参与磷酸化AMKα亚基172位苏氨酸,使AMPK激活[11]。最近研究发现,TAK1为一种MAPKK(促分裂原活化蛋白激酶)激酶-7,在AMPK活化通路中起着重要作用[12]。CaMKK调节AMPK活性并非通过改变AMP/ATP比值起作用,而是通过调节细胞内Ca2+离子浓度,使Ca2+离子浓度升高而激活AMPK[13]。
1.2.2 AMP/ATP比值 AMPK为机体内细胞的主要“能量感受器”,在多种代谢反应中均有所参与。AMPK的活性受到AMP/ATP比值的限制,AMP/ATP比值可调控其活性,当代谢反应时,细胞内的ATP消耗增多或生成减少均会导致AMP/ATP比值增大,进而激活AMPK。ATP的生成减少或者消耗增加通常出现在机体处于缺氧或者低血糖情况下,当AMPK激活时,提供能量给机体。研究表明,AMPK已成为调控AMP/ATP比值水平的“燃料开关”,而AMP/ATP比值也成为激活AMPK的重要条件,使机体的能量代谢处于稳态[14]。
1.2.3 激素或细胞因子 当机体处于静息状态时,脂联素、瘦素、抵抗素、二甲双胍和罗格列酮等也可以激活AMPK。脂联素是一种脂类细胞所分泌的肽类激素,其可有效激活AMPK,而瘦素一般在骨骼肌处起作用,磷酸化AMPKαM亚基而使AMPK被激活[15]。
图1 AMPK的激活及调控[16]
Fig. 1 AMPK activation and regulation
众所周知,肝脏能有效维持机体的能量平衡,尤其是糖脂类的代谢。当饥饿或空腹时,肝脏可通过体内糖原分解或糖异生途径来维持机体内血糖的稳定[17]。肝脏的糖脂代谢功能障碍是肥胖、糖尿病等代谢疾病的基本特征,而以上疾病的主要发病表现为肝脏胰岛素抵抗,其一般生理病理特征为糖原分解、糖异生功能紊乱,进而引起肝脏葡萄糖大量堆积。因此,怎样有效控制糖尿病所致机体肝脏过分糖异生,降低肝脏内葡萄糖的输出,是糖尿病等代谢疾病治疗的一个新的研究方向[3]。
AMPK在机体的能量代谢中起着总开关的作用,AMPK在机体许多脏器中均有表达,研究表明,在机体或者细胞的葡萄糖稳态调节中AMPK发挥着重要作用,已成为研究糖脂代谢的重要靶点[4]。AMPK在肝脏糖脂代谢中发挥着重要作用,主要通过抑制肝脏葡萄糖的生成和脂质的合成发挥作用。在肝脏糖脂代谢中,通过在正常或者胰岛素抵抗的Zuker大鼠中注射AICAR(AMPK激活剂)可抑制肝脏糖异生,从而降低血糖水平;原代培养的肝细胞通过二甲双胍处理能激活AMPK来抑制葡萄糖的生成[18];二甲双胍不能改善LKB1基因敲除小鼠的血糖水平[19]等实验,表明了AMPK在肝脏糖代谢中发挥的重要作用。
在肝脏脂质稳态的调节中,脂肪酸和胆固醇合成过程中的2个关键酶乙酰辅酶A羧化酶(ACC)和3-羟基-3-甲基戊二酰辅酶A还原酶(HMG-COA)是最早发现的AMPK靶蛋白,ACC是脂肪酸合成前体丙二酸单酰辅酶A合成的关键限速酶,AMPK可通过使ACC以及HMG-COA发生磷酸化抑制其活性,从而抑制脂肪酸的合成[20]。另一方面,AMPK也能降低转录因子肝细胞固醇调节元件结合蛋白(SREBP-1C)和碳水化合物反应元件结合蛋白(ChREBP)的活性,从而抑制脂质合成相关基因的表达[21],表明AMPK在肝脏脂质调节过程中的重要作用。
糖异生指的是非糖物质如甘油、乳酸、氨基酸等合成转变成糖原或葡萄糖的反应。机体内一半以上的葡萄糖利用及重大器官的供能均来自于肝脏糖异生。肝脏糖异生除了受胰岛素、胰高血糖素和糖皮质激素的调节,也受一系列转录因子的调控,通过这些转录因子的串话最终把信号反馈到糖异生2个关键性限速酶:葡萄糖-6-磷酸酶(glucose-6-phosphatase,G6Pase)与磷酸烯醇式丙酮酸羧化酶(phosphoenolpyruvate carboxykinase,PEPCK)。以上2种限速酶转录所需的时间决定机体肝脏糖异生的快慢[22]。
3.1 肝脏糖异生关键酶及转录因子
近年来,由于糖异生作用研究的不断深入,对其关键调控基因的探究不断增多。通过不断地深入研究,人们对于肝脏糖异生的基本分子机制也有所完善,发现糖异生在各个作用环节中受到多种转录因子及相关酶的调控。
3.1.1 PEPCK PEPCK是一种糖异生过程中的关键性限速酶,在糖异生反应时草酰乙酸经PEPCK催化脱羧基转变为磷酸烯醇式丙酮酸,这是糖异生反应过程中最先发生且最为重要的反应环节[23]。PEPCK有2种同工酶(isozymes),分别为细胞质里所存在的PEPCK-C及线粒体内所存在的PEPCK-M。PEPCK-C是糖异生反应过程中的一种关键性限速酶,PCK1为PEPCK-C的编码基因,与糖尿病等疾病紧密相关,PCK1如果发生突变,将导致糖异生作用过度,引起肝脏葡萄糖的输出增加[24]。
研究发现,糖尿病db/db模型鼠感染抑制PCK1的RNAi腺病毒后,PEPCK-C mRNA与其相关的蛋白的表达能力均下降,可有效改善糖尿
病等一系列代谢性疾病,肝糖异生反应能有效下调关键基因FOXO1、HNF-4α、PGC-1α的表达,也能控制限速酶G6Pase的转录,并且可除去Sirt1(氧化还原态敏感元件)对于肝糖异生的调控反应。这些为PEPCK-C及其相关编码基因在充当代谢综合性疾病的治疗的新靶点提供了较为有利的依据[25]。 相反,普通小鼠靶向剔除PCK1基因后,PEPCK-C过度表达,诱发肥胖、糖尿病等病患。肝脏中PCK1基因敲除的小鼠,肝脏PEPCK-C水平比正常大约高出7倍,使肝脏糖异生作用显著增强,葡萄糖输出增加[26]。
3.1.2 G6Pase G6Pase也是糖代谢中的关键酶之一。G6Pase是内源性葡萄糖输出最后环节的一个重要限速酶,有直接决定糖异生作用以及机体葡萄糖稳态的重要作用,G6Pase已成为研究糖尿病等代谢性疾病重要靶点。G6Pase在糖尿病生理病理学及病因学中都存在某种程度上的作用,并且对G6Pase的干扰能对糖尿病患者空腹高血糖具有较佳的控制。但通过使用G6Pase靶向治疗糖尿病的研究还在起步阶段,对G6Pase的研究还需深入,期待有新型抗糖尿病药物开发成功[27]。
3.1.3 叉头框/翼螺旋转录因子家族成员 FoxO1与Foxa2为叉头框/翼螺旋转录因子家族中较为重要的2个成员,归类于核受体亚家族,在肝脏中往往被用于脂肪、葡萄糖和血液内胰岛素含量的监测[28]。Fox转录因子家族是Akt的直接作用靶点,能有效调控胰岛素-糖脂的代谢。Foxa2为一种体内脂质代谢的重要作用因子,能有效提升肝脏对于胰岛素的敏感力,降低葡萄糖的生成。Foxa1的主要职能是加快空腹肝糖的异生速度,使肝糖输出量进一步上升[29]。通过研究观测到,一般情况下,胰岛素相关的信号转导途径可调节Foxa1及Foxa2活性,使糖脂代谢、氧化达到平衡。而当胰岛素发生抵抗时,FoxO1被激活,导致糖异生作用增加及高血糖,脂肪酸氧化增加等作用[30]。Foxa1转录因子是胰岛素活性的关键靶点之一。应用转基因研究及腺病毒转染观测到,过量表达Foxa1的实验鼠易出现糖耐受能力损害,肝糖原含量及脂肪沉积量都下降; 而FoxO1功能缺如的实验鼠出现胰岛素敏感力上升,肝脏糖异生受抑的现象,以上均表示FoxO1表达紊乱易引起胰岛素对于肝脏糖脂代谢能力的调控受损[31]。总之,FoxO1被激活,能够增加G6Pase和PEPCK的表达,而FoxO1被抑制,则G6Pase和PEPCK的表达也被抑制,导致糖异生作用被抑制,使得肝脏葡萄糖的产生减少。
3.1.4 过氧化物酶增殖体激活受体γ共激活蛋白1在空腹条件下,肝脏葡萄糖输出是保持机体能量代谢平衡的基础,而肝脏葡萄糖的合成受控于转录调节因子,其中转录辅助激活因子:过氧化物酶增殖体激活受体γ共激活蛋白1家族(peroxisome proliferators-activated receptor-γ coactivator-1,PGC-1)在肝糖脂代谢的反应过程中具有较为重要的意义。PGC-1家族存在2种亚型:PGC-1α及PGC-1β,2种亚型均为应用氨基端的LXXLL保守序列和相关核受体产生交互反应,其相应的保守肽序列失去将引起PGC-1对肉毒碱棕榈酰基转移酶(CPT)及PEPCK的诱导能力下降[32]。相关实验发现,PGC-1α能够激活大鼠肝脏和肝细胞糖异生相关基因的表达,在1、2型糖尿病患者肝脏内,PGC-1α mRNA表达含量上升,PGC-1α可使肝糖异生相应基因HNF-4α与FoxO1的表达有效激活,进而促使肝糖的输出量增多[33]。表明了PGC-1α是肝脏糖异生的关键调控基因之一。
3.1.5 cAMP及其相关辅激活物 cAMP的效应元件结合蛋白(cAMP-response element binding protein,CREB)为重要的核转录因子,是碱性亮氨酸拉链(bZIP)家族成员,CREB调节的转录辅激活因子2(CREB regulated transcription coactivator 2,TORC2)为控制糖异生作用的“分子开关”。TORC2主要表达于肝细胞中,当肝细胞中cAMP上升时,TORC2的表达也上升,使下游PEPCK、G6Pase、PGC-1基因的表达均上升,以上作用可被CREB抑制剂降低,表明TORC2的表达有赖于CREB,TORC2与CREB结合后相应糖异生反应才能开启[34]。所以,抑制TORC2的去磷酸化或禁止其转移至核内,有助于肝糖合成降低。
3.1.6 糖皮质激素受体(glucocorticoid receptor,GR) GR和相关糖皮质激素在核内结合后,与活化的PGC-1α相互作用后影响2个糖异生作用关键酶G6Pase与PEPCK基因的表达,起到对糖异生的调控作用。
3.2 AMPK与肝脏糖异生作用的联系
体内葡萄糖稳态主要由肝脏糖异生以及葡萄糖摄取维持。正常情况下胰岛素也可应用糖异生的调节来调控机体内血糖的含量,但对于2型糖尿病患者,其机体分泌胰岛素不足且存在胰岛素抵制作用,胰岛素抑制肝糖异生的作用紊乱,使得葡萄糖浓度升高,发生高血糖。AMPKα亚基Thr172发生磷酸化激活后,抑制糖异生相关基因PEPCK、G6Pase的表达,抑制肝脏糖异生,从而降低血糖水平[36]。
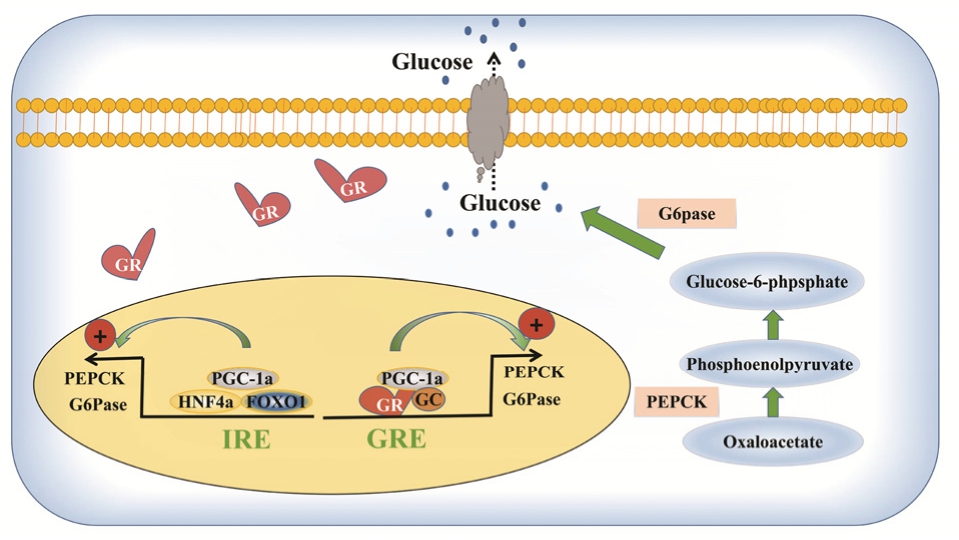
图2 肝糖异生分子调控示意图[35]
Fig. 2 The regulation of hepatic gluconeogenesis
CREB是糖异生调节机制中的一个转录因子,也是调节糖异生通路2种关键限速酶PEPCK、G6Pase的关键转录因子,然而CREB本身不足以转录激活靶基因,主要是通过CREB和CREB结合蛋白(CBP)结合转变为复合体后,对G6Pase及PEPCK 2种关键性基因进行激活,开启糖异生反应[37]。TORC2也可调控CREB的活性。研究表明,禁食期间,胰高血糖素产生量上升刺激TORC2去磷酸化,与CREB相结合并聚集于细胞核内,开启糖异生反应相应基因的转录反应。Yuan等[38]研究表明,被激活的AMPK通过使CREB发生磷酸化而抑制糖异生;Liu等[39]研究表明,AMPK活化可以抑制肝脏糖异生关键转录因子TORC2和FoxO1的表达,AMPK活化通过使TORC2发生磷酸化停滞在细胞质内,使CREB转录降低,抑制糖异生2种关键性酶PEPCK与G6Pase的表达进而控制肝脏糖的异生反应[40]。另外,Horike等[41]研究表明,AMPK活化使糖原合成激酶3β(glycogen synthase kinase 3 beta,GSK-3β)磷酸化,降低CRE的转录活性以及PEPCK的表达,从而抑制肝脏糖异生。
FoxO1主要是应用与G6Pase及PEPCK基因启动子相应胰岛素反应元件(insulin response element,IRE)相结合,有效调节G6Pase与PEPCK 2种关键性酶的表达,从而调节糖异生作用。FoxO1表达紊乱会导致胰岛素调节肝脏糖脂代谢的能力受损。通过研究观测到,PGC-1α在胰岛素功能缺陷的实验鼠肝细胞内具有强烈的诱导作用,进而开启肝脏糖异生反应的关键性酶PEPCK及G6Pase的表达,使肝糖的输出量大大上升,以上反应时,PEPCK的活化需使用糖皮质激素受体GR及肝细胞核因子HNF-4α的协同作用,而后二者通过PGC-1诱导而激活[42]。AMPK可通过增加孤儿核受体(SHP)的表达,抑制CREB与TORC2的结合,也能影响肝细胞核因子4α(hepatic nuclear factor 4 alpha,HNF-4α)及FoxO1的转录活性,从而控制糖异生反应,下降机体空腹血糖[43]。
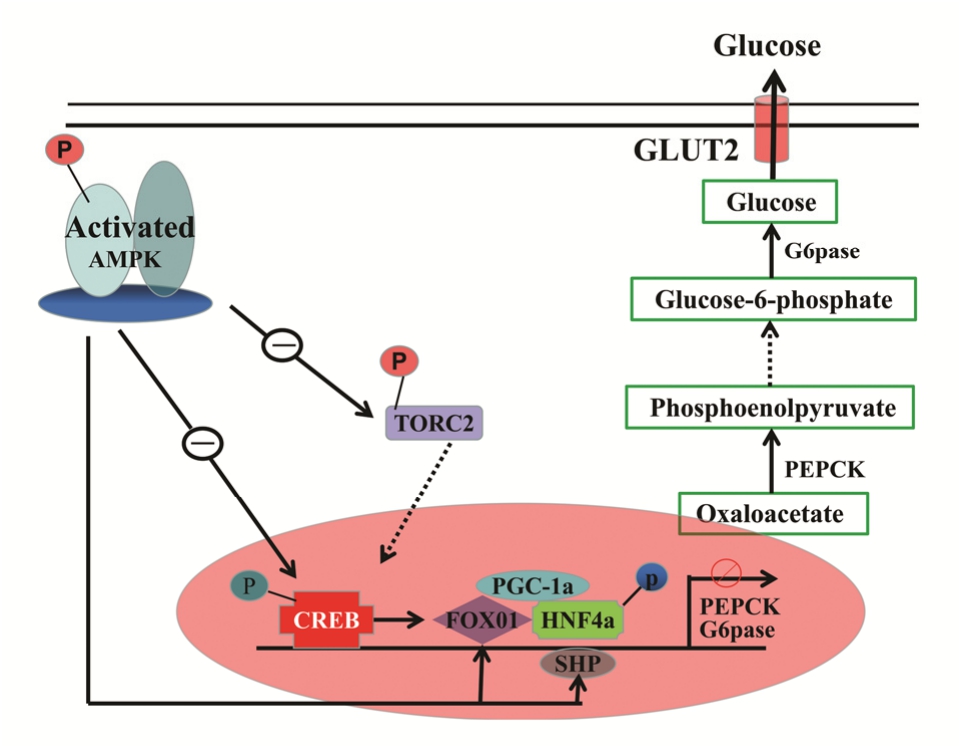
图3 AMPK调节肝糖异生示意图[35]
Fig. 3 The AMPK regulate hepatic gluconeogenesis
AMPK是机体和细胞的“能量感受器”,对机体和细胞的能量稳态起着重要作用。肝脏是机体重要的代谢器官,肝脏糖异生作用又是葡萄糖代谢重要组成部分。AMPK与肝脏糖脂代谢密切相关,其应用抑制肝脏相关糖异生信号通路进而控制葡萄糖的代谢反应。在肝脏的糖异生反应过程中,受到很多激素以及转录因子的调控,如FOXO1、CREB、TORC2、HNF-4α、PGC-1α等。AMPKα亚基在Thr172发生磷酸化激活后,能抑制糖异生酶及其相关转录因子的有效表达,抑制肝脏发生糖异生反应,进而下降血糖含量。总之,AMPK参与肝脏糖脂代谢,与肝脏糖异生信号通路转录因子存在联系,这些对进一步研究AMPK在治疗糖脂代谢紊乱引起的各种疾病有着重要的意义。
REFERENCES
[1] KUANG R, LIU L, LIU Y, et al. Anti-hyperglycemic effect of Fenugreek seed and Mulberry leaf extract formula on the blood glucose levels of the insulin resistance and metabolism disorder rats models and the mechanism [J]. Chin J Mod Appl Pharm(中国现代应用药学), 2016, 33(5): 551-556.
[2] DONG J Z, MA W, LIU Y, et al. Effects of metformin on sugar lipid metabolic disorder caused by sulpiride or risperidone in rats [J]. Chin J Mod Appl Pharm(中国现代应用药学), 2015, 32(1): 17-22.
[3] CHUNG S T, HSIA D S, CHACKO S K, et al. Increased gluconeogenesis in youth with newly diagnosed type 2 diabetes [J]. Diabetologia, 2015, 58(3): 596-603.
[4] KIM Y D , PARK K G, LEE Y S, et al. Met for mininhibits hepatic gluconeogenesis through AMP-activated protein kinase dependent regulation of the orphan nuclear receptor SHP [J]. Diabetes, 2008(57): 306-314.
[5] WEI S, LI W, YU Y, et al. Ginsenoside compound K suppresses the hepatic gluconeogenesis via activating adenosine-5'monophosphate kinase: A study in vitro and in vivo [J]. Life Sci, 2015(139): 8-15.
[6] HARDIE D G. AMP-activated protein kinase: maintaining energy homeostasis at the cellular and whole-body levels [J]. Annu Rev Nutr, 2014(34): 31-55.
[7] GRAHAME HARDIE D. AMP-activated protein kinase: a key regulator of energy balance with many roles in human disease [J]. Intern Med, 2014, 276(6): 543-559.
[8] QIU S L. AMP-activated protein kinase alpha 2 protects against liver injury from metastasized tumors via reduced glucose deprivation-induced oxidative stress [J]. J Biol Chem, 2014, 289(13): 9449.
[9] 葛斌, 谢梅林, 顾振纶, 等. AMPK作为治疗2型糖尿病新靶点的研究进展[J]. 中国药理学通报, 2008, 24(5): 580-583.
[10] SHAW R J, KOSMATKA M, BARDEESY N, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress [J]. Proc Natl Acad Sci USA, 2004, 101(10):3329-3235.
[11] XIE M, ZHANG D, DYCK J R, et al. Apivotal role for endogenous TGF-β-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway [J]. PNAS, 2006, 1039(46): 17378-17383.
[12] HAWLEY S A, PAN D A, MUSTARD K J, et al. Calmodul independent prote in kinase kinse-βis an alternative up stream kinase for AMP-activated protein kinase [J]. Cell Metab, 2005, 2(1): 9-19.
[13] HARDIE D G. AMP-activated prote in kinase as a drug target [J]. Annu Rev Pharmaclo Toxicol, 2007(47): 185-210.
[14] MAGNONI L J , VRASKOU Y, PALSTRA A P, et al. AMPK-activated protein kinase plays an important evolutionary conserved in the regulation of glucose metabolism in fish skeletal muscle cells [J]. PLoS One, 2012, 7(2): e31219.
[15] SCHIMMACK G, DEFRONZO R A, MUSU N. AMP-activated protein in kinase role in metabolism and the rapeuticim placations [J]. Diabetes Obes Metab, 2006, 8(6):591-602.
[16] ZHANG B B, ZHOU G, LI C. AMPK: An emerging drug target for diabetes and the metabolic syndrome [J]. Cell Metabolism, 2009, 9(5): 407-416.
[17] STAEHR P, HOTHER-NIELSEN O, BECK-NIELSEN H. The role of the liver in type 2 diabetes [J]. Rev Endocr Metab Disord , 2004, 5(2): 105-110.
[18] SHAW R J, LAMIA K A, VASQUEZ D, et al. The kinase LKB1 mediates glucosehomeostasisin liver and therapeuticeffects of met formin [J]. Science, 2005, 310(5754):1642-1646.
[19] 王斐. 二甲双胍对高脂饲养大鼠肝脏AMPK和PPARs活性影响-改善脂肪肝的可能机制[D]. 济南: 山东大学, 2008.
[20] LEE H U, BAE E A, HAN M J, et al. Hepatoprotective effect of ginsenoside Rbl and compound K on tert-buty1 hydroperoxide-induced liver injury [J]. Liver Int, 2005, 25(5):1069-1073.
[21] ZHANG P, TU B, WANG H, et al. Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion [J]. Proc Natl Acad Sci USA, 2014, 111(29): 10684-10689.
[22] 韩向晖. 肝脏糖异生的分子机制研究进展[J]. 世界华人消化杂志, 2008 (16): 3659-3665.
[23] BEALE E G, HAMMER R E, ANTOINE B, et al. Disregulated glyceroneogenesis: PCK1 as a candidate diabetes and obesity gene [J]. Trends Endocrinol Metab, 2004(15):129-135.
[24] MÉNDEZ-LUCAS A, DUARTE J A, SUNNY N E, et al. PEPCK-M expression in mouse liver potentiates, not replaces, PEPCK-C mediated gluconeogenesis [J]. J Hepatol, 2013, 59(1): 105-113.
[25] GOMEZ-VALADES A G, MENDEZ-LUCAS A, VIDALALABRO A, et al. Pck1 gene silencing in the liver improves glycemia control, insulin sensitivity, and dyslipidemia in db/db mice [J]. Diabetes, 2008, 57(8): 2199-2210.
[26] BEALE E G, HARVEY B J, FOREST C. PCK1 and PCK2 as candidate diabetes and obesity genes [J]. Cell Biochem Biophys, 2007, 48(2/3): 89-95.
[27] 方显锋. 葡萄糖-6-磷酸酶与糖尿病[J]. 国外医学(内分泌学分册), 2003(5): 305-307.
[28] QU S, ALTOMONTE J, PERDOMO G, et al. Aberrant forkhead box O1 function is associated with impaired hepatic metabolism [J]. Endocrinology, 2006, 147(12): 5641-5652.
[29] ZHANG W, PATIL S, CHAUHAN B, et al. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression [J]. J Biol Chem, 2006, 281(15): 10105-10117.
[30] MAIESE K. FoxO transcription factors and regenerative pathways in diabetes mellitus [J] Curr Neurovasc Res, 2015, 12(4): 404-413.
[31] SADANA P, PARK E A. Characterization of the transactivation domain in the peroxisome-proliferatoractivated receptor gamma co-activator (PGC-1) [J]. Biochem J, 2007, 403(3): 511-518.
[32] LIN J, TARR P T, YANG R, et al. PGC-1beta in the regulation of hepatic glucose and energy metabolism [J]. Biol Chem, 2003, 278(33): 30843-30848.
[33] YOON J C, PUIGSERVER P, CHEN G, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1 [J]. Nature, 2001, 413(6852): 131-138.
[34] XIA X, YAN J, SHEN Y, et al. Berberine improves glucose metabolism in diabetic rats by inhibition of hepatic gluconeogenesis [J]. PLoS One, 2011, 6(2): e16556.
[35] 李伟. 人参皂苷Compound K对2型糖尿病的降血糖作用及肝糖异生信号转导通路调控[D]. 长春: 吉林大学, 2012.
[36] KOO S H, FLECHNER L, QI L, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism [J]. Nature, 2005, 437(7062): 1109-1111.
[37] LE L J, TUTEJA G, WHITE P, et al. CRTC2 (TORC2) contributes to the transcriptional response to fasting in the liver but is not required for the maintenance of glucose homeostasis [J]. Cell Metab, 2009, 10(1): 55-62.
[38] YUAN H D, PIAO G C. An active part of Artemisia sacrorum Ledeb. Suppresses gluconeogenesis through AMPK mediated GSK3β and CREB phosphorylation in human HepG2 cells [J]. Biosci Biotechnol Biochem, 2011, 75(6): 1079-1084.
[39] LIU Y , DENTIN R, CHEN D, et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange [J]. Nature, 2008, 456 (7219): 269-273.
[40] MONTMINY M, KOO S H, ZHANG X. The CREB family:key regulators of hepatic metabolism [J]. Ann Endocrinol (Paris), 2004, 65(1): 73-75.
[41] HORIKE N, SAKODA H, KUSHIYAMA A, et al. AMP-activated protein kinase activation increases phosphorylation of glycogen synthase kinase 3beta and thereby reduces cAMP-responsive element transcriptional activity and phosphoenolpyruvate carboxykinase C gene expression in the liver [J]. J Biol Chem, 2008, 283(49):33902-33910.
[42] OH K J, HAN H S, KIM M J, et al. CREB and FoxO1: two transcription factors for the regulation of hepatic gluconeogenesis [J]. BMB Rep, 2013, 46(12): 567-574.
[43] JIANG S J, DONG H, LI J B, et al. Berberine inhibits hepatic gluconeogenesis via the LKB1-AMPK-TORC2 signaling pathway in streptozotocin-induced diabetic rats [J]. World J Gastroenterol, 2015, 21(25): 7777-7785.
Research Progress of AMPK and Hepatic Glucolipid Metabolism
CHEN Jincheng1,2, ZHANG Yin1*(1.Department of Clinical Pharmacy, the Second Affiliated Hospital of Fujian Medical University, Quanzhou 362000, China; 2.College of Pharmacy, Fujian Medical University, Fuzhou 350108, China)
ABSTRACT:AMP activated protein kinase (AMPK) is a kind of kinase that widely involved in various metabolic regulation, which is the main cellular energy sensor. AMPK is activated by the Thr172 phosphorylation of alpha subunit, which can control lipid synthesis and glucose stability by the inhibition of the related transcription factor in lipid synthesis and hepatic gluconeogenesis. Liver gluconeogenesis is regulated by the hormone and crosstalks among transcription factors such as FOXO1, CREB, TORC2, HNF-4α, PGC-1α and eventually put the signal back to the two key enzyme genes PEPCK and G6Pase of gluconeogenesis. The activated AMPK can inhibit expression of key gluconeogenic enzymes and transcription factors, thereby inhibition of hepatic gluconeogenesis and reducing the blood glucose level. In this review, we aim to discuss and summarize the role of AMPK in liver glucolipid metabolism, focusing on the analysis of the link between AMPK and transcription factors of liver gluconeogenesis signaling.
KEY WORDS:AMP activated protein kinase (AMPK); hepatic glucolipid metabolism; gluconeogenesis; PEPCK; G6Pase; transcriptin regulators
中图分类号:R966
文献标志码:A
文章编号:1007-7693(2017)07-1062-06
DOI:10.13748/j.cnki.issn1007-7693.2017.07.030
引用本文:陈金成, 张吟. AMPK与肝脏糖异生研究进展[J]. 中国现代应用药学, 2017, 34(7): 1062-1067.
收稿日期:2016-10-26
(本文责编:曹粤锋)
基金项目:2013年国家卫生计生委共建科学研究课题(WKJ-FJ-13);2015年福建省卫生系统中青年骨干人才培养项目(2015-ZQN-ZD-23)
作者简介:陈金成,男,药师 Tel: 15859088929 E-mail: 453886460@qq.com*
通信作者:张吟,女,副教授,硕导 Tel:(0595)22775021 E-mail: zyin1973@163.com