 ±s表示,检验水准为α=0.05,所有检验均为双侧检验。因样本量较小,当出现参数检验与非参数检验结果不同时,以非参数检验的结果为准,并用Bonferroni法校正检验水准进行多重比较;2种方法结果相同时,采用参数方法的结论。
±s表示,检验水准为α=0.05,所有检验均为双侧检验。因样本量较小,当出现参数检验与非参数检验结果不同时,以非参数检验的结果为准,并用Bonferroni法校正检验水准进行多重比较;2种方法结果相同时,采用参数方法的结论。
李志峰,邓漫漫,查 洁,周 勇,方志鸿,徐 兵 *(厦门大学附属第一医院血液科,福建 厦门 361003)
摘要:目的 研究新型小分子酪氨酸激酶抑制剂Apatinib对急性髓系白血病(acute myeloid leukemia,AML)干祖细胞增殖和凋亡的影响及其相关分子机制。方法 CCK8法检测不同浓度Apatinib对kg1α细胞的增殖抑制作用,Annexin V/PI法检测不同浓度Apatinib诱导kg1α细胞和原代CD34 +AML干细胞的凋亡情况,Western blot法检测Apatinib处理kg1α细胞后PI3K/AKT通路相关蛋白(AKT、Raf和PTEN)的表达变化。结果 不同浓度的Apatinib(2.5,5,10,20,40 µmol·L -1)作用48 h和72 h后对kg1α细胞均具有显著的增殖抑制作用,呈浓度和时间依赖性,与对照组相比差异均具有统计学意义(P<0.05或P<0.01)。Annexin V/PI检测细胞凋亡的结果显示,不同浓度apatinib对kg1α具有显著的诱导凋亡作用,作用48 h和72 h后的凋亡率和对照组相比差异均有统计学意义(P<0.01)。不同浓度Apatinib作用48 h后对7例原代AML干细胞均具有显著的杀伤作用,与对照组相比差异具有显著的统计学意义(P<0.01);Western blot结果显示,Apatinib处理kg1α细胞48 h后AKT/p-AKT、p-Raf表达降低,而p-PTEN表达增加。结论 Apatinib可抑制AML干祖细胞样细胞kg1α细胞的增殖,且可诱导kg1α细胞和原代CD34 +AML干祖细胞的凋亡,均呈浓度和时间依赖性,其作用机制可能是通过干扰PI3K/AKT通路实现的。
关键词:Apatinib;AML干祖细胞样细胞株(kg1α);PI3K/AKT pathway
急性髓系白血病(acute myeloid leukemia,AML)是成人急性白血病最常见的类型,难治、复发是目前导致AML治疗失败的最根本原因,临床中可分为M0~M7共8种。研究表明,白血病干细胞(leukemia stem cell,LSC)具有自我更新、无限增殖的潜能,多处于细胞静止期(G0期),对传统的化疗药物不敏感,是导致白血病复发及难治的根本原因 [1]。因此对LSC的靶向治疗是AML最根本的治疗策略。
目前很难获得足够研究的LSC,常规的方法是从白血病患者体内提取LSC。有研究表明 [2-3],男性急性微分化型粒细胞白血病(AML-M0)病人来源的Kglα细胞系,其高表达CD34(98%)、CD123(90%),多数不表达CD38,与目前公认LSC的免疫表型特征相一致,且其高表达P糖蛋白对常规AML化疗药物耐药。可作为比较理想的AML干祖细胞样细胞进行相应的研究。
大量研究发现,多种恶性血液病(白血病、淋巴瘤、多发性骨髓瘤等)的发生发展同样与新生血管生成密不可分 [4-5]。新生血管形成有赖于促血管生成因子和抑血管生成因子的精密调控,其中最重要的促血管生成因子是血管内皮生成因子(vascular endothelial growth factor,VEGF),其主要通过与细胞膜表面的血管内皮生长因子受体(vascular endothelial growth factor receptor,VEGFR)结合而促进血管生成 [6]。自从1971年Folkman首次提出肿瘤的生长依赖于新生血管形成,抗血管生成药逐步发展成为一种有效的抗肿瘤治疗手段,且为当今肿瘤研究的主要热点之一。
Apatinib(YN968D1)是我国自主研发的一种选择性抑制VEGFR2的小分子化合物,其亦可抑制RET、c-KIT及c-Src等 [7-8]。目前II/III临床试验结果显示,Apatinib对多种实体瘤如胃癌、肝癌及乳腺癌等有显著杀伤作用 [9-12],而关于其在AML干祖细胞中的作用尚不明确,本实验探讨Apatinib对AML干祖细胞样细胞株kg1α的杀伤作用及其相关机制。
1.1 药物、试剂
甲磺酸阿帕替尼(江苏恒瑞医药股份有限公司);IMDM培养基(Gibco公司);胎牛血清(Hyclone公司);CCK8(日本同仁化工Dojindo公司,批号:JQ701);Annexin-V/APC调亡检测试剂盒(批号:4283875)、抗人CD34(批号:E10158-1635)及CD38抗体(批号:E11027-1634)均购于eBiosecience公司;CD34 MicroBead Kit,human(德国Miltenyi Biotec公司,货号:130-046-703、130-092-263);RIPA裂解液(碧云天公司);兔抗Akt、兔抗p-Akt473及兔抗β-actin单抗购于Cell Signaling Technology (CST)公司。BD公司的FACSArial用于kg1α细胞的分选及周期检测,而BD AccuriC6用于细胞凋亡的检测,Bio Tek公司的Synergy H1多功能酶标仪用于检测CCK8实验的OD值。
1.2 仪器
CO 2培养箱(美国Themo scientific公司);CX4光学显微镜(日本Olympus);5804离心机(德国Eppendorf);Model 550酶标仪(美国Bio-Rad);FACSC6和FACSArial流式细胞仪(美国BD公司)。
1.3 细胞株及其培养条件
在37 ℃、5%CO 2恒温培养箱中,kg1α细胞置于含10%胎牛血清的IMDM培养基中生长,取对数生长期细胞离心,台盼蓝染色并计数,拒染率>98%者适用于实验。
1.4 细胞增殖和毒理试验
取对数生长期细胞悬液以每孔5×10 4接种于96孔板中,分别设置0,2.5,5,10,20,40 μmol·L -1组,每孔100 μL细胞悬液,并加入含10%新生小牛血清的IMDM培养基100 μL作为空白对照组,每个浓度均设3个复孔,培养24 h后再加入CCK8工作液,每孔10 μL,继续在培养箱中培养2~4 h,采用酶标仪测定450 nm处的吸光度值(OD)。细胞抑制率(%)=(1-药物组吸光度值/对照组吸光度值)×100%。
1.5 Annexin V-APC/PI双染法流式细胞仪检测Apatinib处理后kg1α细胞的凋亡
取对数生长期细胞悬液以每孔2×10 5接种于24孔板中,共设置0,10,20,30,40 μmol·L -1剂量组,作用24 h后离心收集细胞,采用PBS洗涤1次,弃上清,加入50 μL binding buffer重新悬浮细胞,再加入0.5 μL Annexin V-APC冰上避光染色30 min,后再用binding buffer洗涤1次,弃上清,再次加入200 μL binding buffer重新悬浮细胞后加入1 μL PI,5 min后上机检测细胞凋亡状态。
1.6 Ficoll淋巴细胞分离液密度梯度分离原代AML单个核细胞及免疫磁珠(Magnetic activated cell sorting,MACS)分选原代CD34 +AML干细胞
将AML患者骨髓液和等体积的PBS充分混匀后,沿着50 mL离心管管壁小心加至含有等体积的Ficoll淋巴细胞分离液中,800×g离心30 min,然后小心吸出中间白色薄膜层,并重悬于等体积PBS中充分混匀,用600×g离心15 min,弃上清,PBS洗涤1次,加入PBS混匀后计数;每1×10 7个细胞加入100 μL PBS和10 μL的CD34-APC抗体,4 ℃避光孵育30 min,加1 mL的PBS洗涤1次,弃上清,加入100 μL的PBS和10 μL抗体APC的免疫磁珠,混匀后4 ℃孵育15~20 min,再用PBS洗涤1次,弃上清,重悬于500 μL分选液,加到磁场LD柱中,收集流出的细胞并计数。
1.7 Annexin V-APC/PI双染法流式细胞仪检测Apatinib处理后原代CD34 +AML干细胞的凋亡
取“1.6”项下分选后原代CD34 +AML干细胞悬液以2×10 5·mL -1接种于24孔板内,设置0,10,20,30,40 µmol·L -1剂量组,处理48 h后离心收集细胞,然后进行凋亡检测,具体步骤同“1.5”项下方法。
1.8 蛋白质印迹法检测PI3K/AKT通路蛋白的变化
设置Apatinib(0,5,10 µmol·L -1)处理48 h后,收集各个不同处理组的细胞,用含有1%苯甲基磺酸(phenyl methanesulfonyl fluoride,PMSF)的RIPA(radio immunoprecipitation assay)裂解液,于冰上裂解细胞15 min,12 000×g 4 ℃离心15 min,取出上清,根据BCA试剂盒说明书测蛋白浓度,沸水浴变性5 min后,取同等量蛋白样品用10% SDS-PAGE凝胶进行电泳,电泳完毕转至PVDF膜。转膜完成后用含5%脱脂奶粉的TBST溶液中室温封闭1 h,然后分别加入兔抗p-Akt(1∶1 000稀释)、兔抗Akt(1∶1 000稀释)、兔抗β-actin (1∶1 000稀释),在4 ℃摇床上孵育过夜;TBST洗涤3次(每次5 min),加入HRP标记的羊抗兔二抗(1∶10 000稀释)室温孵育1 h,TBST重新洗涤3次,膜与化学发光底物反应3~5 min后,暗室中ECL显色,X射线片压片曝光。
1.9 统计学方法
采用SPSS 19.0软件进行统计学分析,多组间均数比较使用多个独立样本的非参数检验或单因素方差法进行统计(方差不齐时考虑Welch校正),方差齐时考虑使用LSD(方差齐性)进行多重比较,不齐时采用Dunnett’s T3方法进行多重比较;计量资料用
±s表示,检验水准为α=0.05,所有检验均为双侧检验。因样本量较小,当出现参数检验与非参数检验结果不同时,以非参数检验的结果为准,并用Bonferroni法校正检验水准进行多重比较;2种方法结果相同时,采用参数方法的结论。
2.1 CCK8法检测Apatinib对AML干祖细胞样细胞株kg1α细胞增殖的影响
结果显示,Apatinib能明显抑制kg1α细胞增殖,呈剂量依赖性,2.5,5,10,20,40 µmol·L -1Apatinib分别作用于kg1α细胞48 h后,各组细胞活性为(92.32±0.82)%、(80.59±4.95)%、(61.75± 0.47)%、(51.51±4.10)%和(26.42±4.20)%,方差分析结果显示,与对照组相比差异均具有统计学意义(P<0.05),48 h的IC 50浓度为(16.98±0.08)µmol·L -1。上述浓度Apatinib分别作用于kg1α细胞72 h后,各组细胞活性为(88.42±2.91)%、(70.83±4.45)%、(47.87±1.59)%、(31.41±3.57)%、(13.26±1.96)%,方差分析结果显示,与对照组相比差异均具有统计学意义(P<0.01),72 h的IC 50浓度为(10.05± 0.08)µmol·L -1。独立样本t检验分析表明10,20,40 µmol·L -1作用72 h细胞活性显著低于48 h,差异均具有统计学意义(P<0.01),提示Apatinib抑制kg1α细胞增殖呈时间依赖性。结果见图1。
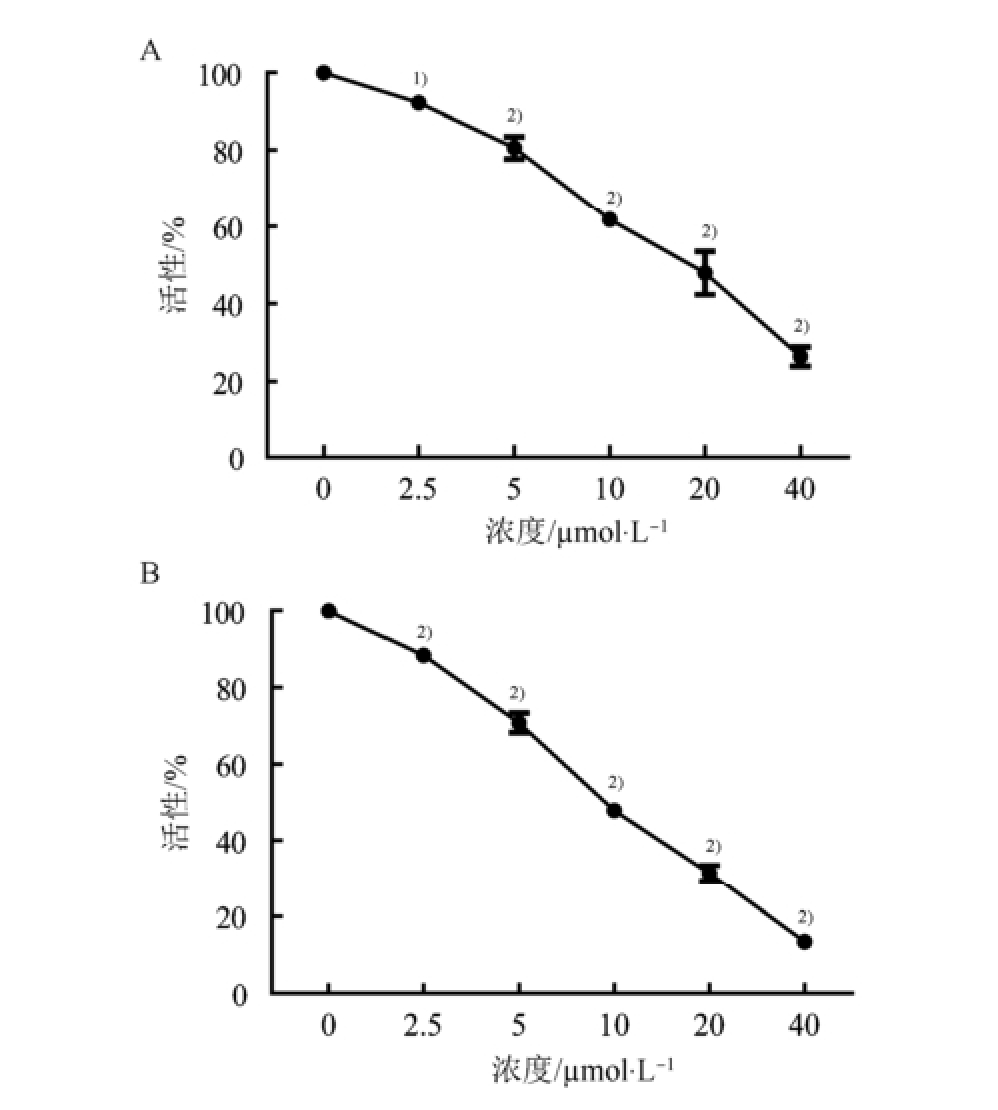
图1 Apatinib作用48,72 h后明显抑制AML干祖细胞样细胞株kg1α细胞的增殖
A-48 h;B-72 h;与对照组(0 µmol·L
-1)相比,
1)P<0.05,
2)P<0.01。
Fig.1 Apatinib significantly inhibited AML stem-like cells kg1α proliferation after treating with 48, 72 h
A-48 h; B-72 h; compared with control group(0 µmol·L
-1),
1)P<0.05,
2)P<0.01.
2.2 流式细胞术检测Apatinib处理AML干祖细胞样细胞株kg1α细胞后的凋亡情况
Annexin V-APC/PI双染法流式细胞术比较不同浓度的Apatinib作用于kg1α细胞48 h和72 h后诱导凋亡的情况。10,20,30,40 µmol·L -1Apatinib作用kg1α细胞48 h后,各组凋亡率分别为(6.58±0.45)%、(12.6±2.34)%、(16.37±4.38)%和(19.77±2.55)%,随浓度增加,凋亡细胞比例逐渐增加,呈剂量依赖性。方差分析表明,不同浓度Apatinib能诱导kg1α细胞凋亡,与对照组(4.57±1.16)%比较差异有统计学意义(F=18.85,P<0.01)。用LSD方法比较不同浓度Apatinib处理的kg1α细胞两两组间的细胞凋亡的差异,结果表明,10 µmol·L -1与20 µmol·L -1、30 µmol·L -1和40 µmol·L -1相比均具有明显的统计学差异(P<0.05),20 µmol·L -1与40 µmol·L -1相比有明显的统计学差异(P<0.01),其余各组间差异无统计学意义。
上述浓度Apatinib作用kg1α细胞72 h后各组凋亡比例为(10.83±2.96)%、(15.38±2.78)%、(24.95±2.70)%和(35.17±1.36)%,凋亡比例随浓度增加而增加,方差分析表明,不同浓度Apatinib能诱导kg1α细胞凋亡,与对照组(5.03±1.11)%比较差异有统计学意义(F=79.425,P<0.01)。用LSD方法比较不同浓度Apatinib处理的kg1α细胞两两组间的细胞凋亡比较的差异结果表明,10 µmol·L -1与20,30,40 µmol·L -1比较均具有统计学意义(P<0.05),20 µmol·L -1与30,40 µmol·L -1比较均具有统计学意义(P<0.01),30 µmol·L -1与40 µmol·L -1比较也具有统计学意义(P<0.01)。30,40 µmol·L -1Apatinib处理72 h后凋亡比例显著高于48 h,差异具有统计学意义(分别为P<0.05和P<0.01)。显示Apatinib对kg1α细胞的杀伤作用还具有时间依赖性。结果见图2。

图2 Apatinib诱导kg1α细胞凋亡
A-48 h后kg1α细胞的凋亡比例;B-72 h后kg1α细胞的凋亡比例;C-典型的凋亡散点图;与对照组(0 µmol·L
-1)相比,
1)P<0.05,
2)P<0.01。
Fig.2 Apatinib induced kg1α cells apoptosis
A-The apoptotic percentage of kg1α after dealing with apatinib for 48 h; B-the apoptotic percentage of kg1α after dealing with apatinib for 72 h; C-flow cytometric analysis; compared with control group(0 µmol·L
-1),
1)P<0.05,
2)P<0.01.
2.3 Apatinib诱导原代CD34 +AML干细胞的凋亡情况
Annexin V-APC/PI双染法流式细胞术检测Apatinib对不同患者来源的CD34 +AML干细胞的凋亡比例。研究结果显示,不同浓度Apatinib(10,20,30,40 µmol·L -1)作用48 h,对7例不同患者来源的CD34 +AML干细胞均有杀伤作用,呈剂量依赖性,凋亡比例分别为(32.83±16.72)%,(41.91± 16.36)%,(51.48±14.99)%,(53.50±15.91)%,与对照组(18.70±11.48)%相比,差异具有显著的统计学意义(F=6.219,P<0.01)。结果见表1和图3。
表1 7例AML患者的临床生物学特征
Tab.1 Clinical biological characteristics of 7 patients
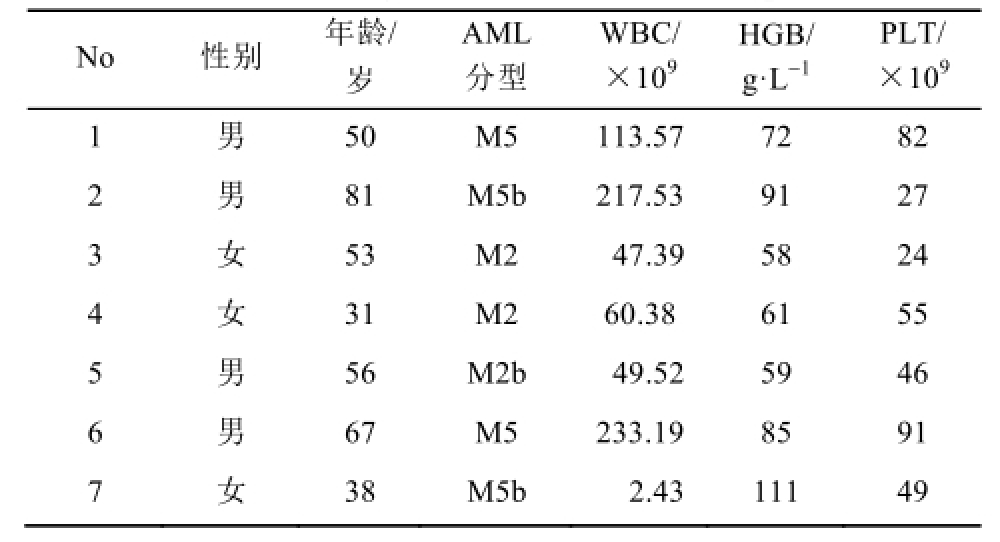
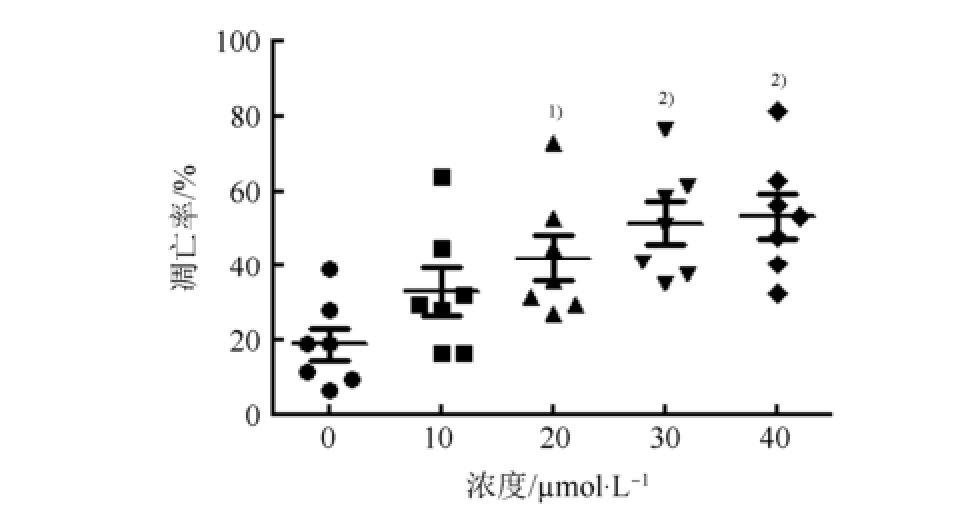
图3 Apatinib对不同来源AML干细胞的杀伤作用
与对照组(0 µmol·L
-1)相比,
1)P<0.01,
2)P<0.001。
Fig.3 The apoptosis effect of Apatinib on AML stem cells derived from different sources
Compared with control group(0 µmol·L
-1),
1)P<0.01,
2)P<0.001.
2.4 Western blot检测Apatinib作用于kg1α细胞后PI3K/AKT通路相关蛋白表达水平的变化
为了阐明Apatinib抑制kg1α细胞的增殖及诱导凋亡的分子机制,Western blot检测不同浓度Apatinib处理kg1α细胞48 h后PI3K/AKT通路相关蛋白的表达变化。数据显示,随着Apatinib浓度的增加,AKT/p-AKT及p-raf表达降低,而PI3K/AKT通路的负调控因子PTEN表达升高。据此推测,Apatinib可通过抑制PI3K/AKT信号通路从而抑制kg1α细胞增殖和诱导其凋亡。结果见图4。

图4 Apatinib作用于kg1α细胞48 h后PI3K/AKT通路相关蛋白的表达变化
Fig.4 Western blot showed that Apatinib could modulatet the PI3K/AKT signaling pathway-related proteins expression after treating with 48 h
急性髓系白血病是一组起源于造血干/祖细胞的恶性血液病,在临床特性、细胞遗传学及分子生物学等方面具有高度异质性 [13]。随着高剂量联合化疗和造血干细胞移植的应用,AML预后有所改善,但难治复发仍是临床面临的一大难题。有研究表明LSC是导致白血病复发及难治的根本原因,而目前治疗模式包括大剂量化疗、甚至造血干细胞移植仍无法完全消灭LSC,因此急需寻求一种杀伤和消灭LSC的新策略。
分子靶向治疗是目前恶性血液病研究领域的热点,是指针对细胞受体、基因、调控分子等信号传导作为靶点的治疗,研究最为广泛的靶向治疗药物主要有EGFR家族抑制剂、血管生成抑制剂、信号传导抑制剂和凋亡诱导剂等 [14-16],其中最成功的典范是酪氨酸激酶抑制剂伊马替尼(Imatinib)治疗慢性髓系白血病,70%~80%患者可获长期生存。相对于传统的化疗药物,分子靶向药物具有作用疗效好,不良反应小等特性,因此,越来越多的学者专注于分子靶向药物的研究。
Apatinib(YN968D1)作为一种新型的小分子酪氨酸激酶抑制剂,是Valaitnib的类似物,通过选择性抑制VEGFR-2活化从而发挥抗肿瘤血管新生。Tian等 [8]证实Apatinib在体外和小鼠肿瘤移植模型中对多种实体瘤(如肺癌、结肠癌、胃癌及肝癌等)具有显著的杀伤作用。同时,Yan等研究发现 [17],Apatinib可逆转P-gp和BCRP等介导的多药耐药作用。目前Apatinib的II/III临床试验结果显示对胃癌、非小细胞肺癌、乳腺癌等具有一定的临床有效性 [9-12]。关于Apatinib对血液肿瘤的作用尚不明确,本研究探讨Apatinib对AML干祖细胞样细胞kg1α的杀伤作用及其分子机制。研究发现Apatinib具有明显的抑制kg1α细胞增殖,呈剂量和时间依赖性,48 h和72 h的IC 50浓度分别为(16.98±0.08)µmol·L -1和(10.05±0.08)µmol·L -1。研究还表明Apatinib能诱导kg1α细胞的凋亡,也表现为浓度和时间依赖性。
受体型酪氨酸激酶(receptor tyrosine kinases,RTKs)广泛存在于体内多种生理过程中,主要通过其下游的多条信号通路(如PI3K/AKT、Src/STAT及RAS/Raf/MEK等)调节细胞的增殖、分化、生存及凋亡等 [18]。大量研究表明,RTKs及其下游通路的激活或发生突变常与肿瘤的发生发展相关,抑制酪氨酸激酶的活性能有效控制肿瘤的生长。本研究显示小分子酪氨酸激酶抑制剂Apatinib作用于kg1α细胞48 h能显著抑制RTKs下游的PI3K/AKT通路蛋白的表达,如AKT/p-AKT,而上调此通路天然负调控因子PTEN的表达。
综上,本研究结果显示在体外Apatinib能抑制kg1α细胞增殖并诱导其凋亡,其作用机制可能是通过抑制酪氨酸激酶活性,进而阻断其下游PI3K/AKT通路的信号传导而发挥杀伤作用。
REFERENCES
[1] GROVE C S, VASSILIOU G S. Acute myeloid leukaemia: a paradigm for the clonal evolution of cancer [J]. Dis Model Mech, 2014, 7(8): 941-951.
[2] DE LEEUW D C, DENKERS F, OLTHOF MC, et al. Attenua-tion of microRNA-126 expression that drives CD34+38-stem/progenitor cells in acute myeloid leukemia leads to tumor eradication [J]. Cancer Res, 2014, 74(7): 2094-2105
[3] LI Y, CHEN K, ZHOU Y, et al. A New strategy to target acute myeloid leukemia stem and progenitor cells using chidamide, a histone deacetylase inhibitor [J]. Curr Cancer Drug Targets, 2015, 15(6): 493-503.
[4] CHAND R, CHANDRA H, CHANDRA S, et al. Role of microvessel density and vascular endothelial growth factor in angiogenesis of hematological malignancies [J]. Bone Marrow Res, 2016(2016): 5043483. doi: 10.1155/2016/5043483.
[5] IMAI N, SHIKAMI M, MIWA H, et al. t(8;21) acute myeloid leukaemia cells are dependent on vascular endothelial growth factor (VEGF)/VEGF receptor type2 pathway andPhosphorylation of Akt [J]. Br J Haematol. 2006, 135(5): 673-682.
[6] ELLIS L M, HICKLIN D J. VEGF-targeted therapy: mechanisms of anti-tumour activity [J]. Nat Rev Cancer, 2008, 8(8): 579-591.
[7] PENG H, ZHANG Q, LI J, et al. Apatinib inhibits VEGF signaling and promotes apoptosis in intrahepatic Cholangiocarcinoma [J]. Oncotarget, 2016, 7(13): 17220-17229.
[8] TIAN S, QUAN H, XIE C, et al. YN968D1 is a novel and Selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potent activity in vitro and in vivo [J]. Cancer sci, 2011, 102(7): 1374-1380.
[9] AOYAMA T, YOSHIKAWA T. Targeted therapy: Apatinib-New third-line option for refractory gastric or GEJ cancer [J]. Nat Rev Clin Oncol, 2016, 13(5): 268-270.
[10] LI J, QIN S, XU J, et al. Randomized, double-blind, placebo-controlled phase iii trial of apatinib in patients with chemo-therapy refractory advanced or metastatic adenocarcinoma of the stomach or gastroesophageal junction [J]. Am J Clin Oncol, 2016, 34(13): 1448-1454.
[11] GENG R, LI J. Apatinib for the treatment of gastric cancer [J]. Expert Opin Pharmacother, 2015, 16(1): 117-122.
[12] HU X, ZHANG J, XU B, et al. Multicenter phase II study of apatinib, a novel VEGFR inhibitor in heavily pretreated patients with metastatic triple-negative breast cancer [J]. Int J Cancer, 2014, 135(8): 1961-1969.
[13] ESTEY E, DOHNER H. Acute myeloid leukaemia [J]. Lancet, 2006, 368(9550): 1894-907.
[14] ZANIBONI A, FORMICA V. The best first anti-EGFR before anti-VEGF, in the first-line treatment of RAS wild-type Meta-Static colorectal cancer: from bench to bedside [J]. Cancer Chemother Pharmacol, 2016, 78(2): 233-244.
[15] LI K, LI J. Current molecular targeted therapy in advanced gastric cancer: a comprehensive review of therapeutic mechanism, clinical trials, and practical application [J]. Gastroenterol Res Pract, 2016(2016): 4105615. doi: 10.1155/ 2016/4105615.
[16] EMOLE J, TALABI T, PINILLA-IBARZ J. Update on the management of philadelphia chromosome positive chronic myelogenous leukemia: role of nilotinib [J]. Biologics, 2016(10): 23-31.
[17] MI Y J, LIANG Y J, HUANG H B, et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters [J]. Cancer Res, 2010, 70(20): 7981-7991.
[18] REGAD T. Targeting RTK Signaling Pathways in cancer [J]. Cancers(Basel), 2015, 7(3): 1758-1784.
Apatinib Inhibits Proliferation and Induces Apoptosis of Acute Myeloid Leukemia Stem/progenitor Like Cell Line (kg1α cells) and Its Mechanism
LI Zhifeng, DENG Manman, ZHA Jie, ZHOU Yong, FANG Zhihong, XU Bing *(Department of Hematology, The First Affiliated Hospital of Xiamen University, Xiamen 361003, China)
ABSTRACT:OBJECTIVE To investigate the effect of Apatinib, a small-molecule vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor, on the proliferation and apoptosis of acute myeloid leukemia(AML) stem/progenitor cells and its molecule mechanism. METHODS The kg1α cells and primary CD34 +AML stem cells were treated with a serial of concentrations of Apatinib for 48 h and 72 h, the inhibitory ratio was measured by CCK8 assay, the apoptosis percent was measured by flow cytometry. Western bolt was used to analyzed AKT/p-AKT, p-Raf and p-PTEN expression after treatment with 0, 10, 20 µmol·L -1Apatinib in kg1α cells. RESULTS After treatment with a serial of Apatinib (2.5, 5, 10, 20, 40 µmol·L -1) on AML stem-like cell line(kg1α) for 48 h and 72 h, the cell proliferation were significantly inhibited in a dose- and time-dependent mode. All the differences had statistical significance compared with control group. The results of Annexin V/PI showed that various concentration of Apatinib induced significantly apoptosis on kg1α cells. After 48 h and 72 h, all the apoptosis percentage were significantly higher than control group(P<0.01); Apatinib significantly induced apoptosis of 7 cases primary AML stem cell, the difference has statistical significance; Western blot results indicated decrease the expression of AKT/p-AKT and p-Raf, however, upregulated the level of p-PTEN. CONCLUSION Apatinib can inhibit the proliferation of AML stem/progenitor like cell line(kg1α cells) and also induce the apoptosis of kg1α and primary CD34 +AML stem cells. Its mechanism may be related with the PI3K/AKT signal pathway.
KEY WORDS:Apatinib; acute myeloid leukemia stem/progenitor like cell (kg1α); PI3K/AKT pathway
中图分类号:R965.2
文献标志码:A
文章编号:1007-7693(2017)02-0204-06
DOI:10.13748/j.cnki.issn1007-7693.2017.02.012
收稿日期:2016-07-11
作者简介:李志峰,男,硕士,副主任医师 Tel: (0592)2137255 E-mail: lzf_xm@163.com *
通信作者:徐兵,男,博士,主任医师 Tel: (0592)2137254 E-mail: xubingzhangjian@126.com