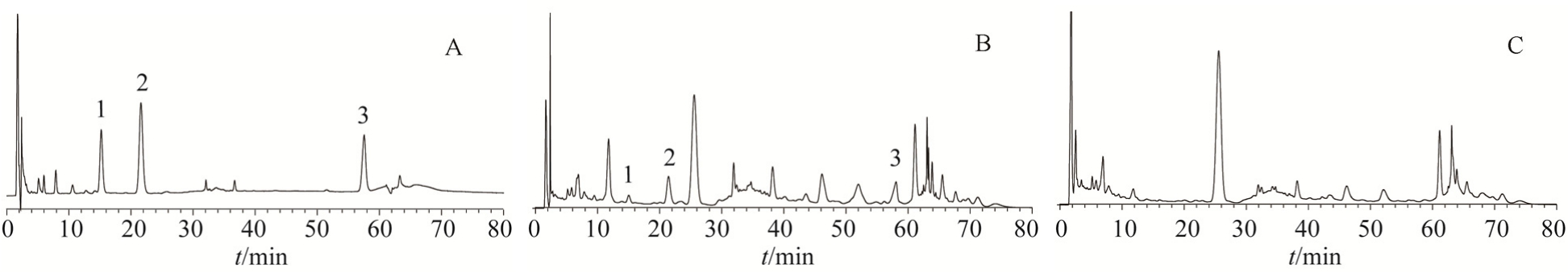
图1 百贝益肺胶囊HPLC色谱图
A-对照品;B-样品;C-阴性样品;1-三七皂苷R1;2-人参皂苷Rg1;3-人参皂苷Rb1。
Fig. 1 HPLC chromatogram of Baibei Yifei capsule
A-control sample; B-sample; C-negative sample; 1-notoginsenoside R1; 2-ginsenoside Rg1; 3-ginsenoside Rb1.
孙荣飞1,张金玺1,赵晓星1,者从章2
(1.昭通市食品药品检验所,云南 昭通 657000;2.永孜堂制药有限公司,云南 昭通 657000)
摘要:目的 增加评价指标,建立科学、合理的百贝益肺胶囊含量测定方法。方法 C18小柱纯化样品,优化色谱条件为:Vp-ODS色谱柱(150 mm×4.6 mm,5 μm);柱温:20 ℃;变动流速;流动相:乙腈(A)-水(B),梯度洗脱;进样量:10 μL;检测波长:203 nm。结果 所测成分三七皂苷R1、人参皂苷Rg1、人参皂苷Rb1分别在40.24~241.44(r=0.999 6),91.50~549.00(r=0.999 9),35.72~241.32(r=0.999 8)μg·mL-1内线性关系良好;加样回收率为 94.15%~99.81%,RSD 为 1.12%~1.94%。结论 该方法准确,重复性良好,可为修订标准提供依据。
关键词:百贝益肺胶囊;含量测定;改进
百贝益肺胶囊始收载于《卫生部颁药品标准》中药成方制剂第 1册,由白及、浙贝母、桔梗、百部、百合、海浮石、紫菀、三七、功劳木、甘草组成,2012年升级成国家标准,增加贵细药材三七的含量测定,以人参皂苷Rg1为评价指标[1]。三七为药典收录品种,中国药典2015年版以3个成分控制其质量[2]11-12;原标准[1]以单一成分,也非标志性成分作为评价指标,存在一定的不科学性,且需要特定Inertsll ODS-SP型色谱柱。因此,本实验增加三七皂苷R1和人参皂苷Rb1为含量测定指标,选择通用ODS短柱,进行方法改进,使其测定方法更加科学合理。
LC-2010AHT高效液相色谱仪(日本岛津公司);MS105DU 电子天平(梅特勒-托利多上海公司);WF-120EH超声仪(宁波海曙五方);WondaSep C18小柱(日本岛津,500 mg/6 mL)。
三七皂苷 R1、人参皂苷 Rg1、人参皂苷 Rb1对照品均来源于中国药品生物制品检定所,批号分别为:110745-200312、110703-201128、110704-201223,含量分别为未标示(以100%计)、93.4%和95.9%;乙腈为色谱纯,甲醇为分析纯,水为纯化水。百贝益肺胶囊(昭通永孜堂制药有限公司,批号:20160520,20161129,20170222,规格:每粒0.4 g);缺三七阴性对照样品(永孜堂制药有限公司,批号:20170222)。
Vp-ODS 色谱柱(150 mm×4.6 mm,5 μm);柱温:20 ℃;流速:0~25 min,1 mL·mim-1,25~30 min , 1 → 0.7 mL·mim-1, 30~80 min ,0.7 mL·mim-1;流动相:乙腈(A)-水(B),梯度洗脱:0~25 min,19%A,25~30 min,19%→28%A,30~65 min,28%→29%A;65~75 min,29%→40%A;75~80 min,40%→19%A);进样量:10 μL。检测波长:203 nm。
2.2.1 对照品溶液的制备 精密称取三七皂苷R1、人参皂苷 Rg1、人参皂苷 Rb1对照品适量,加甲醇分别制成0.80,1.83,0.71 mg·mL-1的储备溶液,即得。
2.2.2 供试品溶液的制备 精密称取本品内容物2.5 g,置于具塞锥形瓶中,精密加入甲醇25 mL,称定重量,超声处理(功率 250 W,频率 40 kHz)30 min,放冷,称定重量,用甲醇补足减失的重量,摇匀,过滤,精密量取续滤液10 mL,蒸干,残渣加水2 mL,使溶解,使通过已活化的C18小柱,依次用水5 mL、20%乙醇10 mL洗脱,弃去洗脱液,再用50%乙醇20 mL洗脱,收集洗脱液,浓缩至近干,转移至10 mL量瓶中,加甲醇至刻度,摇匀,用0.45 μm滤膜过滤,即得。
2.2.3 阴性溶液的制备 精密称取缺三七阴性对照样品2.5 g,按“2.2.2”项下方法制成阴性对照溶液。
2.3.1 专属性试验 分别精密吸取对照品溶液、样品溶液、阴性对照溶液各10 μL,注入液相色谱仪测定。结果供试品溶液色谱中,在与三七皂苷R1、人参皂苷 Rg1、人参皂苷 Rb1对照品相同保留时间处有吸收峰,而阴性样品溶液在相同保留时间处未显吸收峰,表明阴性样品对测定无干扰,结果见图 1。
图1 百贝益肺胶囊HPLC色谱图
A-对照品;B-样品;C-阴性样品;1-三七皂苷R1;2-人参皂苷Rg1;3-人参皂苷Rb1。
Fig. 1 HPLC chromatogram of Baibei Yifei capsule
A-control sample; B-sample; C-negative sample; 1-notoginsenoside R1; 2-ginsenoside Rg1; 3-ginsenoside Rb1.
2.3.2 线性关系 分别取“2.2.1”项下对照品储备液0.5,1.0,1.5,2.0,2.5,3.0 mL置于10 mL量瓶中,加甲醇至刻度,摇匀,进样测定。以峰面积为纵坐标(Y),浓度为横坐标(X),绘制3个对照品的标准曲线。结果三七皂苷 R1回归方程为Y=2 655.91X+203.93(r=0.999 6)、人参皂苷Rg1为Y=2 128.79X+6 143.73(r=0.999 9)、人参皂苷Rb1为Y=2 669.56X+1 2947.70(r=0.999 8),线性范围分 别 为 40.24~241.44, 91.50~549.00, 35.72~214.32 μg·mL-1。
2.3.3 仪器精密度 取同一混合对照品溶液,按“2.1”项下色谱条件连续进样6次,测得三七皂苷 R1、人参皂苷 Rg1、人参皂苷 Rb1的峰面积RSD(n=6)分别为0.6%,0.3%,0.8%,表明仪器精密度良好。
2.3.4 稳定性 取“2.2.2”项下供试品溶液,按“2.1”项下色谱条件,分隔0,2,4,8,24,48 h分别进样,测得混合对照品及供试品中三七皂苷R1、人参皂苷Rg1、人参皂苷Rb1的峰面积RSD(n=6)分别为1.5%,1.0%,1.3%,表明冷藏密封放置48 h,该溶液稳定。
2.3.5 重复性 精密称取同一批号(20160520)样品,按“2.2.2”项下方法平行制备 6份供试品溶液,按“2.1”项下色谱条件进行测定,记录测定峰面积并计算。结果每粒含三七皂苷 R1、人参皂苷 Rg1和人参皂苷Rb1含量的RSD分别为1.9%,1.6%和2.1%(n=6)。
2.3.6 加样回收率 精密称取已知含量的样品1.25 g,加入“2.2.1”项下对照品储备液适量,挥去甲醇,按“2.2.2”项下方法制备成供试品溶液,测得3个成分的峰面积,计算回收率,结果见表1。
2.3.7 样品测定 取样品 3批,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱进样测定,记录峰面积,并计算样品含量,结果见表2。
三七主成分的测定、提取方法相对成熟,基本上类似。水饱和正丁醇反复萃取、大孔树脂分离纯化最为常用[3-6];固相萃取也有运用[7],还有复方丹参片中直接用70%甲醇提取[1]1214-1215。在本实验中,除了比较上述的提取方法,还考察了以下方法:①分别用 10%乙醇、乙醚超声,弃提取液,残渣用甲醇或水饱和正丁醇超声提取;②过氧化铝柱、氧化铝与大孔树脂混合柱,用不同浓度乙醇洗脱;③通过薄层色谱法纯化。从实验结果看,水饱和正丁醇萃取、大孔树脂纯化、固相萃取、薄层色谱纯化可以大幅度去除杂质,其中薄层色谱纯化的样品最好,考虑到实验的简便、准确、重复性,最终选择固相萃取纯化的样品,结合梯度洗脱、流速变化、低柱温的色谱条件,取得相对满意的结果。
表1 三七皂苷R1、人参皂苷Rg1、人参皂苷Rb1回收率
Tab. 1 Recoveries of notoginsenoside R1, ginsenoside Rg1and ginsenoside Rb1

表2 样品含量测定结果(n=3)
Tab. 2 Results of sample determination(n=3) mg

本实验考察了 ZORBAX SB-C18和 Inertsil ODS-3色谱柱,取得了一致的结果。本实验中,柱温对色谱峰的影响明显,短柱在25 ℃以下可以获得良好分离度,35 ℃以上目标峰不能完全分离,结合色谱柱承受温度及分离度,确定柱温为20 ℃,取得良好效果。
[1] 国家食品药品家督管理局发布. 国家药品标准[S]:WS-10115(ZD-0115)-2002-2012Z.
[2] 中国药典. 一部[S]. 2015: 11-12, 1214-1215.
[3] YANG H, LAO G Y, TANG D Z. Determination of notoginsenoside R1, ginsenoside Rg1and ginsenoside Rb1in Compound Huanggen granules by HPLC [J]. China Pharm(中国药师), 2015, 18(1): 55-58.
[4] HUANG Y Y. Determination of contents of notoginsenoside R1, ginsenoside Rg1and ginsenoside Rb1in Sanqi Shangyao capsules by HPLC [J]. China Pharm(中国药房),2015, 26(12): 1706-1708.
[5] ZHANG C H, WANG Y L, FU C. Simultaneous determination of notoginsenoside R1, ginsenoside Rg1and ginsenoside Rb1in Diede pills by HPLC-ELSD [J]. Chin J Mod Appl Pharm(中国现代应用药学), 2016, 33(1): 91-93.
[6] YU X, LIU J, WANG Y N, et al. Simultaneous determination of notoginsenoside R1, ginsenoside Rg1, ginsenoside Re and ginsenoside Rb1in Dredge powder by HPLC [J]. J Shenyang Pharm Univ(沈阳药科大学学报), 2016, 33(7): 537-541.
[7] ZHANG H J, YUAN R Q, HU J Y. Determination of saponins in Formulated Danshen tablets by SPE-HPLC-MSn technique [J]. Lishizhen Med Mater Med Res(时珍国医国药),2011, 22(9): 2174-2176.
Improvement of Content Determination of Baibei Yifei Capsule
SUN Rongfei1, ZHANG Jinxi1, ZHAO Xiaoxing1, ZHE Congzhang2(1.Zhaotong Institute of Food and Drug Control,Zhaotong 657000, China; 2.YongZitang Pharmaceutical Company Limited, Zhaotong 657000,China)
ABSTRACT:OBJECTIVE To augment evaluation index, and establish a scientific and reasonable method for the content determination of Baibei Yifei capsule. METHODS Samples were purified by C18column, optimized chromatographic conditions: Vp-ODS column (150 mm×4.6 mm, 5 μm); column temperature: 20 ℃; fluctuan flow rate; the mobile phase acetonitrile (A)-water (B), gradient elution. The injection volume was 10 μL. The detection wavelength was 203 nm. RESULTS The contents of notoginsenoside R1, ginsenoside Rg1, ginsenoside Rb1were linear in 40.24-241.44(r=0.999 6), 91.50-549.00(r=0.999 9), 35.72-241.32(r=0.999 8)μg·mL-1, respectively. The recovery rate was 94.15%-99.81%, RSD was 1.12%-1.94%.CONCLUSION The method is accurate and reproducible, which can provide the basis for the revision of the standard.
KEY WORDS:Baibei Yifei capsule; content determination; improvement
REFERENCES
中图分类号:R917.101
文献标志码:B
文章编号:1007-7693(2017)12-1740-03
DOI:10.13748/j.cnki.issn1007-7693.2017.12.021
引用本文:孙荣飞, 张金玺, 赵晓星, 等. 百贝益肺胶囊含量测定的方法改进[J]. 中国现代应用药学, 2017, 34(12): 1740-1742.
作者简介:孙荣飞,男,主管药师 Tel: (0870)3156089 E-mail: 286707498@163.com
收稿日期:2017-06-26
(本文责编:曹粤锋)