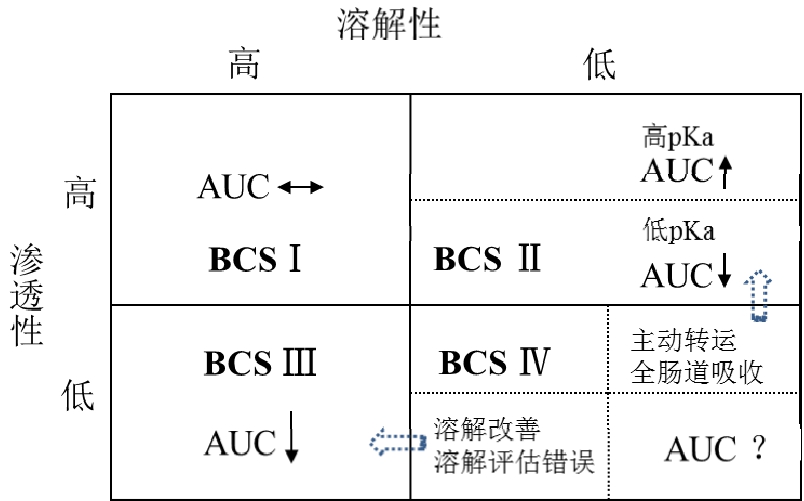
图1 食物对BCS分类药物的影响趋势预测
Fig. 1 Prediction of the influence of food on BCS drugs
·综 述·
臧洪梅,程开生
(安徽医科大学药学院,合肥 230032)
摘要:目的 综述近年来国内外溶出度试验方法的研究进展,为餐后生物利用度的预测提供理论依据。方法 综述药物的食物效应、溶出介质、溶出方法等对溶出度试验体内外相关性的影响。结果 药物性质、食物效应、溶出介质、溶出装置均影响溶出度方法的体内外相关性,均影响口服药物的餐后生物利用度。结论 在一致性评价任务重,国内餐后生物利用度研究经验少的前提下,开发能够预测餐后体内药动学的体外溶出方法,可以一定程度上减少研发风险和成本。
关键词:生物利用度;餐后;溶出度
为规范仿制药质量和疗效一致性评价(以下简称“一致性评价”)工作,国家食品药品监督管理总局2016年发布了《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》,对于口服调释和常释制剂,一般要求进行空腹和餐后生物等效性研究[1]。而中国药典2015年版四部附录《药物制剂与人体生物利用度和生物等效性试验指导原则》[2]对常释制剂一般只要求空腹试验;2005年颁布的《化学药物制剂人体生物利用度和生物等效性研究技术指导原则》也仅明确了空腹生物等效试验的具体要求[3]。由此可见监管部门对仿制药质量与疗效的要求日趋严格,力求从多维度确保仿制药质量和疗效与原研药一致。
对于仿制药是否应进行餐后生物等效研究,在“人用药品注册技术要求国际协调会议”(International Conference on Harmonization,ICH)成员国之间也未达成一致[4],中国药典委员会[2]和欧洲人用医药产品委员会(Committee for Medicinal Products for Human Use,CHMP)[5]认为空腹是检测制剂间潜在差别最敏感的条件,进行餐后生物等效性研究会增加制药企业的法规负担;美国食品药品监督管理局[6]认为食物对生物利用度有影响,因此对于仿制药的要求是,除非说明书指出应空腹服用,否则均应进行餐后生物等效性研究。
食物可能通过如下方式改变生物利用度[7]:延迟胃排空、刺激胆汁分泌、改变胃肠道(GI)pH值、增加内脏血流量、与制剂或药物发生物理和化学反应等。通常,餐后即刻服药,食物对生物利用度影响最大。食物的营养成分、热量、体积和温度能改变胃肠道的生理环境,由此影响药物/制剂滞留时间、溶解度、渗透性进而影响生物利用度。事实上,如果没有特殊的研究手段,很难阐明食物影响生物利用度的机制,尤其是生物药剂学分类系统(bioavailability classification system,BCS)中Ⅱ、Ⅲ、Ⅳ类药物的速释制剂和缓控释制剂,因为食物对其影响非常复杂。在国内一致性评价任务重、餐后生物等效研究经验少、临床基地数量少的情况下,为了提高餐后生物等效试验的成功率,有必要开发能够预测餐后体内药物动力学的体外溶出方法,以减少研发的风险和成本。本文综述药物性质与食物效应、溶出介质、溶出装置、数据评估方法的研究进展,旨在为餐后生物利用度的预测提供理论依据。
根据药物的溶解性和渗透性不同,BCS将口服吸收药物分为 BCS Ⅰ(溶解度高,渗透性好)、BCS Ⅱ(溶解度低,渗透性好)、BCS Ⅲ类(溶解度高,渗透性差)、BCS Ⅳ(溶解度低,渗透性差)4类。食物对生物利用度的影响可根据药物的生物药剂学分类特点作出经验性判断,见图1。Gu等[8]统计了92组临床数据,发现食物对67%的BCS Ⅰ药物无影响,食物对71% BCS Ⅱ药物有正效应,对61% BCS Ⅲ药物有负效应,对73% BCS Ⅳ药物有正效应。对于BCS Ⅱ类药物,根据其pKa、酸碱性、亲脂性可预测出食物的影响[9]。对于高pKa的药物,食物促进胆汁分泌,提高酶活性,提高溶出,且药物解离型比例降低,有利于跨膜吸收,因而有正效应;对于低 pKa药物,进食后胃和小肠上端pH上升,药物解离型比例升高,不利于跨膜吸收,因而产生负效应。食物对于BCS Ⅲ药物产生负效应可能源自吸收面积改变、竞争性吸收和食物的综合作用造成的跨膜吸收的减少。食物对于BCS Ⅳ类药物的影响比较复杂,例如采用制剂技术手段提高了BCS Ⅳ类药物的溶解度,食物对其影响表现为BCS Ⅲ类药物的特点。还有一些 BCS Ⅳ类的药物采用体外小肠渗透试验判定虽然不符合高渗透性定义,但是在整个胃肠道吸收很好或有主动转运吸收,此时食物对其影响就表现为BCS Ⅱ类药物的特点。而对于采用制剂学手段造成药物的体内释放改变或者有特异性吸收窗的药物,很难预测其食物效应。
图1 食物对BCS分类药物的影响趋势预测
Fig. 1 Prediction of the influence of food on BCS drugs
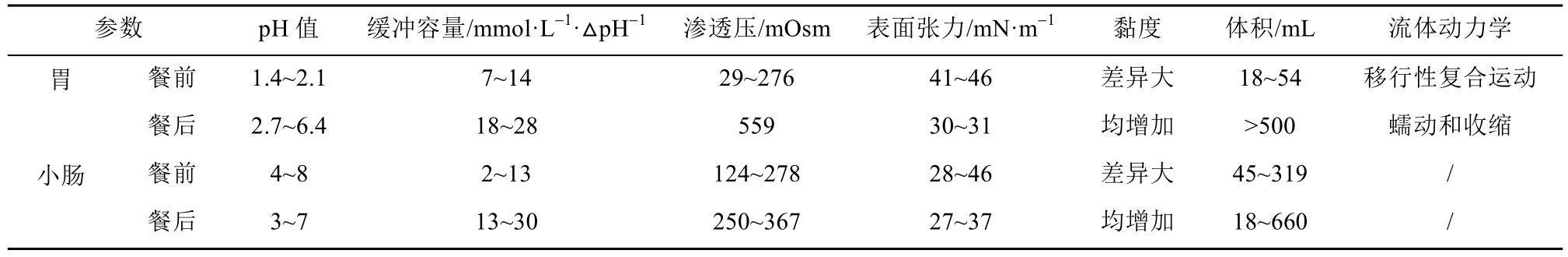
建立体内外相关性好的体外溶出方法用于餐后生物利用度的预测,需要优选出能模拟胃肠液体组成和特性的生理相关性溶出介质,而胃肠液的组成和特性会受食物影响,发生pH值、渗透压、表面张力等参数的改变[10],餐前、餐后胃肠道生理环境差异见表1。
溶出度试验的常用介质主要有水及不同 pH值的缓冲液,仅能反映胃肠液内的pH值变化,不能体现胃肠道生理环境的实际变化,如渗透压、电离能力、黏度、表面张力等参数的变化。近年来,研究者建立了一些能更好地反映空腹和进食状态下胃液和肠液的组成及生理特点的生物相关性溶出介质。
表1 餐前、餐后胃肠道生理环境差异
Tab. 1 Differences of physiological environment of gastrointestinal tract before and after meal
Vertzoni等[11]建立了含有胃蛋白酶和少量胆盐及卵磷脂的模拟空腹状态下的人工胃液(fasted state simulating gastric fluid,FaSSGF),其组成见表2,通过加入生理浓度的胃蛋白酶降低表面张力,与美国药典采用的含有十二烷基硫酸钠的模拟胃液相比更为合理。对比酮康唑在人体胃液、犬胃液、FaSSGF、HCl溶液(pH 分别为 1.6,1.8,2.9)等介质中的溶解度,FaSSGF与人体胃液最接近。
根据进食后胃液的组成和生理参数,Kalantzi等[12]在牛奶的基础上建立了模拟进食后胃液的介质(fed state simulating gastric fluid,FeSSGF)。FeSSGF模拟了大部分进食后胃内产生的生理变化,除了很好地模拟餐后胃内pH值、胆酸盐水平的升高外,缓冲容量、渗透压、表面张力的变化也得到了很好的体现,可以作为溶出介质模拟进食后药物的溶出情况,其组成见表2。
Marques[13]提出了模拟空腹状态下肠液的介质(fasted state simulating intestinal fluid,FaSSIF)-V1,包含胆盐和卵磷脂,这些成分能增加药物的润湿性,同时增加脂溶性药物的溶解。Jantratid等[14]进行了改进,提出了FaSSIF-V2。
Galia等[15]开发了模拟进食状态下肠液的介质(fed state simulating intestinal fluid,FeSSIF)。Jantratid等[14]进行优化,建立了FeSSIF-V2,它反映了餐后pH值、缓冲容量、渗透压和胆汁成分浓度的变化,同时加入了脂类水解产物——甘油单酯,促进难溶性药物的溶出。进食时肠液组成复杂,研究者对FeSSIF的组成难以达成一致,其体内外相关性水平也低于FaSSIF。因此FeSSIF的研究尚待进一步深入,以提高其体内行为的预测能力。
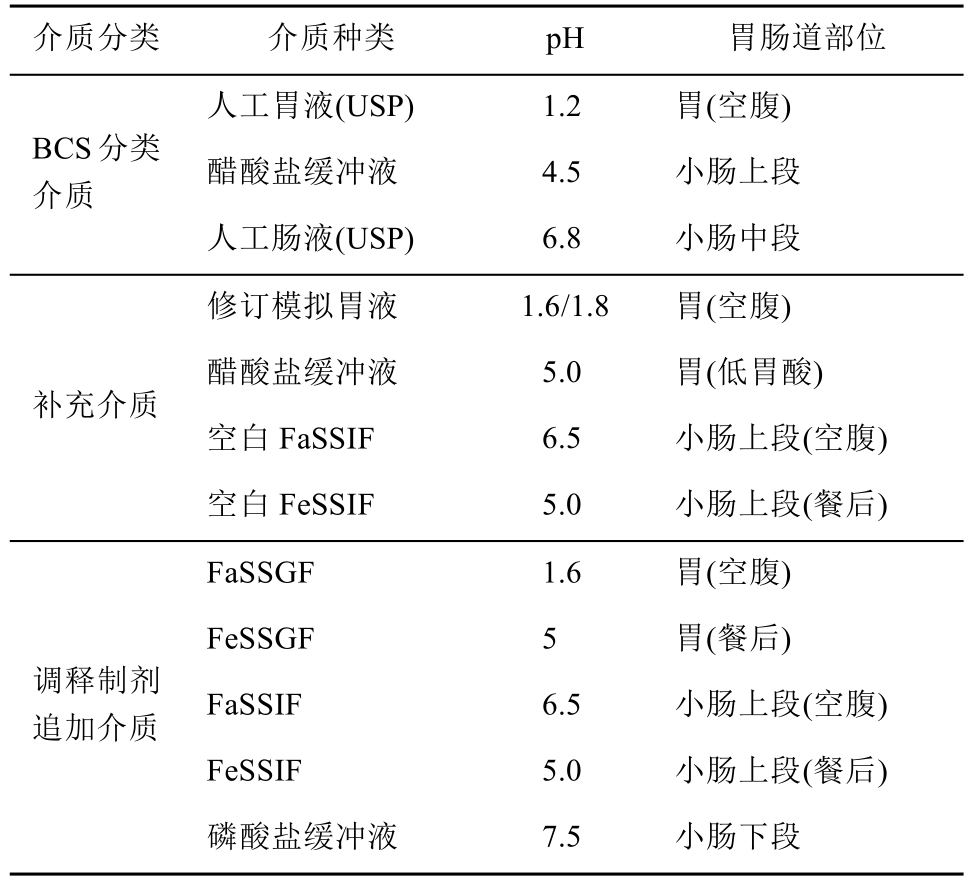
因此应当测定药物在不同介质中的溶解度,对于速释制剂要求测定上消化道相关 pH介质溶解度即可,而调释制剂应当追加小肠下端(pH 7~7.5)和近端结肠部位(pH 5~6)环境下的溶解度[16],还需要增加餐后环境溶解度测试,结果见表3。
溶解度测试后需按BCS分类法计算剂量/溶解度比值,在各pH值下比值<250 mL,则溶出不存在问题;若有在相关介质中比值>250 mL,溶出存在制约因素;若比值在250~1 000 mL,可以继续测试在调释制剂追加介质中的溶解度。尤其是一些亲脂性药物,疏水性高难以润湿的情况下,介质中加入胆酸盐可以克服上述问题而提高溶解度。如果比值>1 000 mL,胆酸盐增溶效果欠佳,可采用其他增溶手段,如表面活性或有机溶剂。
表2 模拟空腹和进食状态下胃液及肠液组成与性质
Tab. 2 Differences of the composition and properties of gastric fluid and intestinal fluid before and after meal

表3 根据药物性质选择合适的溶解度测试介质
Tab. 3 Selecting the appropriate solubility test media according to the nature of the drug
各国药典收载的溶出装置大致 4种:篮法、桨法、往复筒法和流通池法,目前还有多种改进装置正在研究[17]。传统的篮法、桨法装置成本低,应用广泛,其中桨法还能模拟胃肠道内的剪切力,受到研究者的关注较多,并加以改进,如在介质中加入聚苯乙烯小球,模拟药物受胃肠道内容物的摩擦。而流通池法提供的流体力学特性与体内相近,可用来评估制剂在pH值改变或在不同生理相关溶出介质中的溶出行为,可以持续提供溶出所需的漏槽条件,介质可以层流状态冲洗药物,还可以自由更换介质、自动化取样,并且可以在线分析,在难溶性药物研究中应用非常广泛。
Robertson[18]以已上市的 BCS Ⅱ类药品体内药动学特征文献报道为基础,采用模拟胃肠道生理环境的生物相关溶出介质,以开放流通池装置开发溶出测定方法,建立了良好的体内外相关性模型,并应用在创新性药物AMG853(酸性药物)、化合物 A和 AMG221(碱性药物)的处方开发与工艺放大研究中。Medina等[19]发现采用USP收载的桨法无法区分卡马西平片的 4种仿制药和原研制剂的差异,而采用流通池溶出测试则发现 4种仿制药与原研制剂的体外释放特征均不一致。体外酯解模型在脂质制剂(如固体脂质纳米粒)的体外释放中应用广泛,它包括pH值及消化酶的活性控制系统,模拟空腹和餐后小肠内环境,也可用于BCS Ⅱ类药品的食物影响预测[20]。
生理药动学模型(physiologically-based pharmacokinetic,PBPK)以机体的生理学性质参数和药物相关的性质参数为基础建立数学模型,主要应用于化合物筛选、安全性评价、人体药动学规律预测、给药剂量预测、特殊人群(儿童、孕妇)和病理状态下机体的药动学研究以及动态模拟药物-药物相互作用等[21]。Zhang等[22]采用基于PBPK模型的软件 GastroPlus预测了弱碱性低溶解性药物Compound X [pKa 6.2;logP>4;溶解度 300 µg·mL-1(pH 2),1 µg·mL-1(pH 6.8),23 µg·mL-1(FASSIF);190 µg·mL-1(FESSIF)]的食物效应。PBPK 模型可以预测辅料的影响[23],甚至能区分解释结构相近、同为 BCS Ⅱ弱碱性药物酮康唑和泊沙康唑食物效应差异的原因[24]。Heimbach等[25]更进一步结合基于药物体内分布的生物药剂学分类系统(Bio-pharmaceutics drug disposition classification system,BCS/BDDCS)建立了PBPK模型预测食物效应。
在餐后生物等效性试验之前,对药物进行粗略的 BCS分类对于结果的预测有很好的指导作用,但是这种分类比较简单、机械,实际上药物溶解度随所处环境的pH值变化而变化,这样一个药物就会有多种BCS类别,因此有必要结合药物的酸碱性(pKa值)、溶解度等进行综合分析。根据药物能够电离出氢离子的能力可以将药物分为酸性药物、碱性药物和中性药物。多数药物在不同的pH值环境下以不同的解离程度存在,因此具有不同的溶解度、渗透和吸收特性[26]。进食后,胃肠液的pH值、渗透压、表面张力等参数会发生变化,因此食物对各类药物的吸收会产生不同的影响。为了准确预测药物的餐后生物等效性,必须建立体内外相关性高的溶出度方法。首先要参考药物性质与食物效应的关系,然后选择合适的溶出介质及溶出方法,必要情况下参考药动学模型的评估,才能获得良好的体内外相关性。以下将综合文献信息对上述方法的可行性进行验证。
中性药物多为亲脂性化合物,在水溶液中溶解度较低,其吸收特性受胃肠道 pH值变化影响小,但是进食后胃肠液表面张力及渗透压的变化可能导致其吸收速度和程度发生变化。达那唑是炔孕酮的甾类类似物,用于治疗子宫内膜异位,遗传性血管水肿等病症。达那唑溶解性差(37 ℃,0.42 µg·mL-1),脂溶性强(log P 4.53),在胃肠道渗透性好,是典型的BCS II类药物。Galia等[15]用桨法、人工肠液(美国药典方法)、FaSSIF和FeSSIF 3种介质研究单剂量达那唑的体外溶出情况,结果发现,达那唑在人工肠液(美国药典方法)中几乎不溶出,而在 FeSSIF中的溶出量是 FaSSIF的 3~4倍,提示食物对该药物吸收具有正效应。Charman等[27]在11名健康女性中研究证实,进食后,达那唑的生物利用度提高了近 4倍。这一结果显示,选用FaSSIF和FeSSIF生理相关性介质作为溶出介质可反映 BCS Ⅱ类中性化合物(如达那唑)的体内溶出,较好预测体内外相关性。
弱酸性药物受表面活性剂及胃肠道的 pH值变化影响,在中性或碱性环境下溶解度有所增加,因此吸收受表面活性剂及胃肠道 pH值变化的影响较大。苯妥英为亲脂性弱酸药物,典型的 BCSⅡ类药物,其速释制剂采用不含胆酸盐的药典介质,不论pH值如何变化,在4 h内溶出均<20%,而采用FaSSIF(pH 6.8)、FaSSIF(pH 5.0)时,溶出分别达到 36%和 50%,人体口服餐后生物利用度提高了45%,在新西兰白兔中提高41.7%,此结果与两者溶出提高比例相似[20]。
Robertson[18]以模拟肠介质 SIF采用流通池法开放模式比较AMG 853片剂湿法制粒和直接压片处方体外溶出,发现 2种处方释放达到最高浓度的时间一致,湿法制粒比直接压片的最高浓度高约20%,2 h累积溶出高约50%,预测2种制剂体内行为存在明显差异。猴体内药动学验证了以上预测结果,两处方达峰时间一致,湿法制粒的峰浓度和AUC分别为直接压片的3倍和4倍。优选处方再次采用FaSSIF和FeSSIF生理相关性介质溶出,发现FeSSIF在条件下溶出远低于FaSSIF,预测食物对生物利用度有不利影响,人体药动学实验证实,空腹和餐后 AUC分别为 335,114 h·ng·mL-1。以上结果原因可能在于其溶解度受pH值影响,餐后肠内pH值下降,溶解度也下降,导致生物利用度降低。随后研究者继续开发了其钠盐,其溶出在FaSSIF和FeSSIF释放中一致。
碱性药物在空腹胃液中溶解度好于酸性药物,但是其体内行为却更难预测,因为其溶解度受pH值影响,而个体差异、生理差异导致其溶解情况差异较大,进而体内行为差异大。评价这类药物生物等效性时,体内研究的受试者最好入选服用质子泵抑制剂/H2拮抗剂者或老年人,这些人群空腹时胃内pH>5才能获得区分力。伊曲康唑是亲脂性弱碱药物,在pH>2时,溶解度低于检测限,因此上市的普通胶囊生物利用度<20%,且受食物影响,因此要求同服酸液降低胃液pH值,增加溶解度或高脂餐增溶[28]。而制成羟丙基倍他环糊精包合物后,溶解度和溶出速率大幅提高,在pH<1.2时均>5 mg·mL-1,但是表现在体外溶出中的差异更大;pH 1.8和2.0时相对于pH 1.2时溶出更少,溶出量降至40%和25%;pH升至5时,溶出量仅约为 8%。由于伊曲康唑的口服制剂吸收均不完全,FDA溶出数据库中采用0.1 mol·L-1HCl或不含酶的模拟胃液(pH 1.2)作溶出介质,可能高估了其体内生物利用度,没有区分力,如采用含酶SIF(pH 6.8)可能低估了其体内生物利用度,区分力太强。采用改进的含酶SGF(pH 2.0)及FaSSIF和FeSSIF生理相关性介质可较好预测其体内行为[20]。
采用 SGF-SIF作为溶出介质进行弱碱性化合物 A[18]的体外溶出试验,结果显示处方中是否加入富马酸对溶出结果无影响,累积溶出 20%,beagle犬体内药动学研究表明,各处方生物利用度无差异。但是,当溶出介质采用SIF溶液时,加入富马酸的处方释放完全,提示吸收部位的微酸环境能促进体内释放。
综上所述,要选择合适的溶出条件去预测药物的餐后生物利用度,首先要充分了解药物在不同生理 pH值下的溶解性和胃肠道渗透性,根据BCS分类系统确定类别,再结合药物的亲水亲脂性和pKa,选择合适的释放介质、溶出装置,必要情况下可采用 PBPK模型,充分利用已有文献的药物理化性质与药动学参数进行食物效应预测,并在体外或动物体内进行初步验证,从而降低一致性评价工作中餐后生物等效研究失败风险。
[1] 国家食品药品监督管理局. 以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则[EB/OL]. [2016-03-18]. http://www.sda.gov.cn/WS01/CL1751/ 147583. html
[2] 中国药典. 四部 [S]. 2015: 附录356-362.
[3] SHI X. Interpretation of the technical guidelines for the bioequivalence study of generic chemical drugs with pharmacokinetics parameters as the final evaluation index [J].Shanghai Med Pharm J(上海医药), 2016, 37(7): 16-17.
[4] GARCíA-ARIETA A, GORDON J. Bioequivalence requirements in the European Union: critical discussion [J].AAPS J, 2012, 14(4): 738-748.
[5] Committee for Medicinal Products for Human Use (CHMP).Guideline on the Investigation of Bioequivalence, January 20,2010 [EB/OL]. London: European Medicines Agency (EMA).
[6] U.S. Department of Health and Human Services, Food and Drug Administration. Center for Drug Evaluation and Reserach (CDER). Guidance for Industry. Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA. Draft. Guidance [EB/OL]. 2013.http:www.fda.gov/downloads/drugs/guidancecomplianceregul atoryinformation/guidances/ucm377465.pdf.
[7] U.S. Department of Health and Human Services, Food and Drug Administration. Center for Drug Evaluation and Reserach (CDER). Food-Effect Bioavailability and Fed Bioequivalence Studies [EB/OL]. 2002. http://www.fda.gov/cder/guidance/index.html.
[8] GU C H, LI H, LEVONS J, et al. Predicting effect of food on extent of drug absorption based on physicochemical properties[J]. Pharm Res, 2007, 24(6): 1118-1130.
[9] LENTZ K A. Current methods for predicting human food effect [J]. AAPS J, 2008, 10(2): 282-288.
[10] FU L N, ZHENG J Q, ZHENG G G, et al. Progress in research of bio-relevant dissolution media [J]. Chin Pharm J(中国药学杂志), 2013, 48(24): 2084-2088.
[11] VERTZONI M, DRESSMAN J B, BUTLER J, et a1.Simulation of fasting gastric conditions and its importance for the in vivo dissolution of lipophilic compounds [J]. Eur J Pharm Biopharm, 2005, 60(3): 413-417.
[12] KALANTZI L, GOUMAS K, KALLORAS V, et a1.Characterization of the human upper gastrointestinal contents under conditions simulating bioavailability/bioequivalence studies [J]. Pharm Res, 2006, 23(1): 165-l76.
[13] MARQUES M. Dissolution media simulating fasted and fed states [J]. Dissolution Technologies, 2004, 11(2): 16.
[14] JANTRATID E, JANSSEN N, REPPAS C, et a1. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update [J]. Pharm Res, 2008, 25(7):1663-1676.
[15] GALIA E. Physiologically based dissolution tests [D].Frankfurt: Johann Wolfgang Goethe University, 1999.
[16] KLEIN S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug [J]. AAPS J, 2010, 12(3):397-406.
[17] FU X F, KE X. Recent evolvement of oral drug dissolution technology and in vitro and in vivo correlation [J]. Pharm Clin Res(药学与临床研究), 2012, 20(2): 142-147.
[18] ROBERTSON V K. Applications of a biorelevant in vitro dissolution method using USP apparatus 4 in early phase formulation development [J]. Mol Pharm, 2013, 7(5):1466-1477.
[19] MEDINA J R, SALAZAR D K, HURTADO M, et al.Comparative in vitro dissolution study of carbamazepine immediate-release products using the USP paddles method and the flow-through cell system [J]. Saudi Pharm J: PJ, 2014,22(2): 141-147.
[20] RAMAN S, POLLI J E. Prediction of positive food effect:Bioavailability enhancement of BCS class II drugs [J]. Int J Pharm, 2016, 506(1-2): 110-115.
[21] JIN Y W, MA Y M. Progress in methodology of establishing physiologically based pharmacokinetic models [J]. Acta Pharm Sin(药学学报), 2014, 49(1): 16-22.
[22] ZHANG H, XIA B, SHENG J, et al. Application of physiologically based absorption modeling to formulation development of a low solubility, low permeability weak base:mechanistic investigation of food effect [J]. AAPS Pharm Sci Tech, 2014, 15(2): 400-406.
[23] ChOW E C, TALATTOF A, TSAKALOZOU E, et al. Using physiologically based pharmacokinetic (PBPK) modeling to evaluate the impact of pharmaceutical excipients on oral drug absorption: sensitivity analyses [J]. AAPS J, 2016, 18(6):1500-1511.
[24] CRISTOFOLETTI R, PATEL N, DRESSMAN J B.Differences in food effects for 2 weak bases with similar BCS drug-related properties: what is happening in the intestinal lumen [J]. J Pharm Sci, 2016, 105(9): 2712-2722.
[25] HEIMBACH T, XIA B, LIN T, et al. Case studies for practical food effect assessments across BCS/BDDCS class compounds using in silico, in vitro, and preclinical in vivo data [J]. AAPS J,2013, 15(1): 143-158.
[26] ZHENG S F, GUO W B, WANG Y, et al. Improved dissolution method of loratadine tablets [J]. Chin J Mod Appl Pharm(中国现代应用药学), 2015, 32(2): 178-181.
[27] CHARMAN W N, ROGGE M C, BODDY A W, et al. Effect of food and a monoglyceride emulsion formulation on danazol bioavailability [J]. J Clin Pharmacol, 1993, 33(4): 381-386.
[28] YANG L J, ZHU Y G. Pharmacokinetics and bioequivalence of itraconazole capsules in healthy volunteers after high fat diet [J]. Chin J Pharm(中国医药工业杂志), 2009, 40(7):507-509.
Progress in Prediction of Postprandial Bioavailability of Solid Oral Preparations by Dissolution Test
ZANG Hongmei, CHENG Kaisheng
(School of Pharmacy, Anhui Medical University, Hefei 230032, China)
ABSTRACT:OBJECTIVE To provide a theoretical basis for the prediction of postprandial bioavailability test by in vitro dissolution test in recent years. METHODS In this paper, the effects of food , the dissolution medium and dissolution methods on the in vitro and in vivo correlation of dissolution test were reviewed. RESULTS The food effects of drug, the dissolution medium, dissolution methods and data evaluation methods had effects on the in vitro and in vivo correlations of dissolution methods and the postprandial bioavailability of oral drugs. CONCLUSION In China, there is heavy workload in consistency evaluation and few studies on postprandial bioavailability, the development of an in vitro dissolution method for predicting in vivo pharmacokinetics can reduce the research risk and cost of development.
KEY WORDS:bioavailability; postprandial; dissolution
REFERENCES
中图分类号:R944.9
文献标志码:B
文章编号:1007-7693(2017)12-1768-06
DOI:10.13748/j.cnki.issn1007-7693.2017.12.028
引用本文:臧洪梅, 程开生. 溶出度试验预测固体制剂餐后生物利用度的研究进展[J]. 中国现代应用药学, 2017, 34(12):1768-1773.
基金项目:安徽省自然科学基金项目(1508085QH196)
作者简介:臧洪梅,女,硕士,副教授 Tel: (0551)65161176 E-mail: zangweiwei2005@126.com
收稿日期:2017-04-06
(本文责编:蔡珊珊)