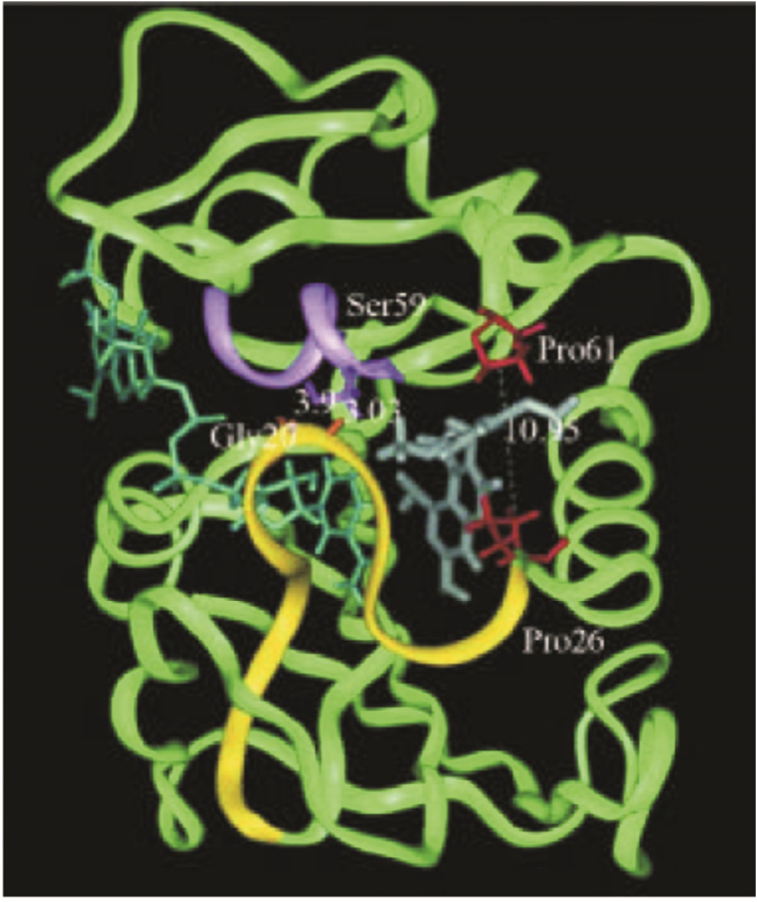
图1 人类二氢叶酸还原酶二级结构
Fig. 1 The secondary structure of human dihydrofolate reductase
李丹1,杨丽君2,翁勤洁2,3*
(1.国家食品药品监督管理总局药品审评中心,北京 100038;2.浙江大学药学院,杭州 310058;3.浙江大学药物安全评价研究中心,杭州 310058)
摘要:二氢叶酸还原酶(dihydrofolate reductase,DHFR)是合成DNA的必需前体物胸腺嘧啶脱氧核苷(dTMP)的关键酶,在细胞增殖中起重要作用,是肿瘤治疗的重要靶点。抑制DHFR能够抑制dTMP的生物合成,进而抑制肿瘤细胞的生长或增殖,在临床上显示抗肿瘤的作用。近年来,该类药物的神经毒性极大限制了其临床广泛应用。基于此,本文对 DHFR抑制剂药物在肿瘤治疗中的应用进行综述,并介绍药物相应的神经毒副作用(药物剂量限制性不良反应),探讨 DHFR抑制剂药物在肿瘤治疗中的发展现状及为临床合理用药减少神经毒性反应提供指导。
关键词:二氢叶酸还原酶(DHFR);抑制剂;抗肿瘤;神经毒性
二氢叶酸还原酶(dihydrofolate reductase,DHFR)基因位于第5号染色体的q11.2-q13.3区域,其编码的 DHFR酶相对分子质量为 2.2×104,由186个氨基酸构成,其等电点为7.7。酶分子的高级结构为中空桶状,桶内侧有几个弹性环,是酶的功能区,晶体结构显示当 DHFR抑制剂接近酶活性中心时,酶分子几个环的构象先后发生变化,从而有利于催化反应进行,桶状结构为底物结合提供了空间定向作用,见图1[1]。DHFR对正常血细胞的生成具有促进作用,对于胚胎发育也是非常重要的,它的缺乏会导致脊椎动物先天性心脏缺损,同时,它的异常表达也是一些肿瘤发生、发展的原因。
DHFR是生物体内叶酸代谢途径的重要酶,在烟酰胺腺嘌呤二核苷酸磷酸(triphosphopyridine nucleotide,NADPH)的存在下催化二氢叶酸还原为四氢叶酸[2]。四氢叶酸是一碳单元的传递体,为合成DNA生物合成所必需的前体物dTMP的过程提供一碳单元。5,10-亚甲基四氢叶酸(5,10-Methylenetetrahydrofolate,Me-THF)经胸苷酸合成酶(thymidylate synthetase,TYMS)的催化生成二氢叶酸,二氢叶酸随后在 DHFR的催化作用下生成四氢叶酸。此外,Me-THF也可通过丝氨酸羟甲基转移酶(serine hydroxymethyrltransferase,SHMT)的催化由二氢叶酸生成或者在亚甲基四氢叶酸脱氢酶(methylenetetrahydrofolate dehydrogenase,MTHFD)的作用下由甲基四氢叶酸生成。另外一个叶酸辅因子,甲酰四氢叶酸由甲酰四氢叶酸合成酶催化二氢叶酸生成或由具有甲川四氢叶酸环水解酶活性的MTHFD催化5,10-甲川四氢叶酸生成,见图2。甲酰四氢叶酸在嘌呤的从头合成中必不可少,是一碳单位的供体,与DNA的从头合成和细胞内的叶酸代谢密切相关。
图1 人类二氢叶酸还原酶二级结构
Fig. 1 The secondary structure of human dihydrofolate reductase

图2 DNA合成及叶酸代谢信号通路
Fig. 2 The signaling pathway of DNA synthesis and folate metabolism
DHFR抑制剂药物的结构与其底物相类似,在肿瘤细胞快速增长过程中,DHFR抑制剂能选择性地与 DHFR结合,阻止或抑制正常底物与酶的结合,抑制其催化还原活性,使二氢叶酸不能转变成四氢叶酸,阻碍叶酸代谢,干扰DNA和蛋白质的合成,最终导致细胞死亡,从而达到治疗肿瘤的目的。
根据化学结构可以将DHFR抑制剂分为2大类:经典DHFR抑制剂和非经典DHFR抑制剂。结构中含有谷氨酸残基的 DHFR抑制剂药物称为经典 DHFR抑制剂。该类药物需要经过细胞膜上的还原叶酸载体蛋白(reduced folate carrier,RFC)的载运进入细胞,随后在叶酸多聚谷氨酸合成酶(folypolyglutamatesyn-thetase,FPGS)的催化下多聚谷氨酸化(Polylutamation),多谷氨酸形式能够增加药物分子滞留细胞的时间,延长药物作用细胞的时间,使细胞内药物浓度增加,同时增强与DHFR的结合性,最终增强其对 DHFR的抑制作用。其中具有代表性的药物为甲氨蝶呤(methotrexate,MTX)和培美曲塞(Pemetrexed,LY231514,商品名:Alimta),结构式见图3。
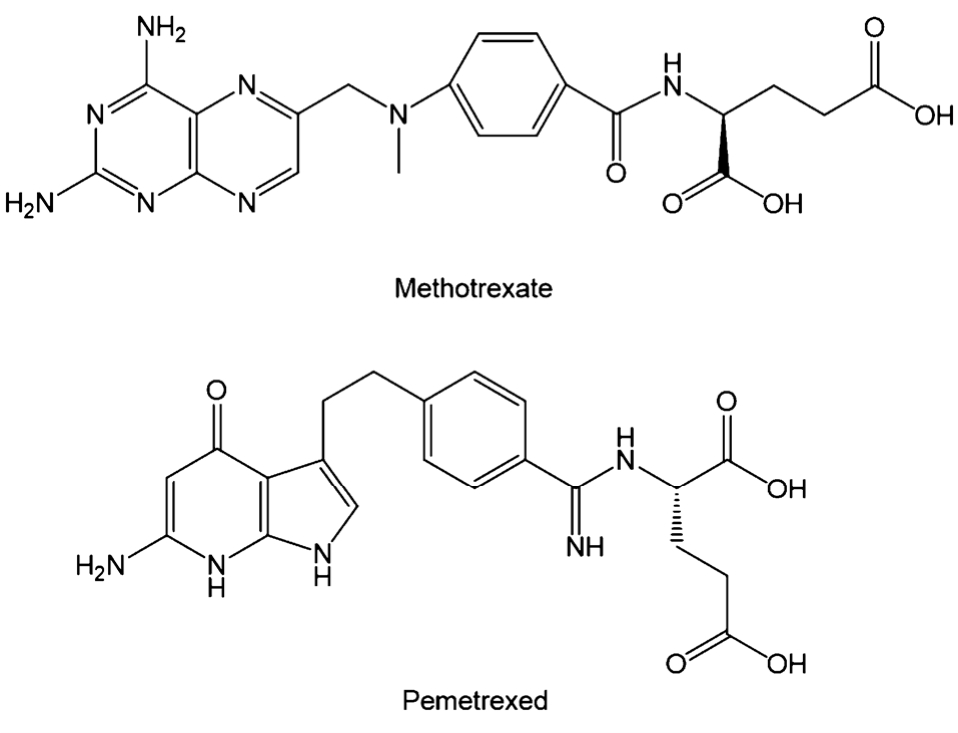
图3 代表性经典DHFR抑制剂的结构
Fig. 3 Representative of the structure of the classic DHFR inhibitors
MTX是第一个有效治疗急性淋巴细胞白血病和治愈绒毛膜癌的药物,是十分有效的抗叶酸药物,竞争性抑制DHFR,目前在临床广泛应用,主要适用于急性白血病、乳腺癌、绒毛膜上皮癌、骨肿瘤等多种肿瘤。同时,MTX也可与其他药物联合运用,治疗乳腺癌、头颈部癌症、骨肉瘤、脑膜炎以及鞘内注射用于治疗中枢神经系统的急性淋巴细胞白血病等。据文献报道,应用MTX治疗之后,使骨肉瘤的复发率、转移率明显下降,同时患者生存率上升至75%[3]。
培美曲塞是一种新型抗叶酸靶点抑制剂药物,属于多靶点抑制剂,不仅作用于DHFR,还作用于 TYMS、甘氮酰胺核糖核苷酸甲酰基转移酶(N-N-N-ribose ribonucleotide formyl transferase,GARFT),因此被称为多靶点抗叶酸剂(multitargetedantifolate,MTA)。培美曲塞使得细胞周期 S期阻滞,抑制肿瘤细胞生长。培美曲塞于2004年 2月被美国食品与药物管理局(U.S. Food and Drug Administration,FDA)批准用于恶性胸膜间皮癌的一线治疗,同年10月被 FDA批准用于晚期非小细胞肺癌(non-small cell lung cance,NSCLC)的二线治疗,在临床实验中显示出广谱抗肿瘤活性,包括结肠癌、胰腺癌、乳腺癌、肝癌、膀胱癌和宫颈癌等。同时与其他药物联合应用,也表现出良好的治疗效果,如培美曲塞联合吉非替尼能够抑制 EGFR突变的非小细胞肺癌对 TKI类的耐药现象[4],并且培美曲塞治疗恶性胸膜间皮癌第Ⅲ期临床试验显示,在Ⅲ期或Ⅳ期患者占78%时,联合组病灶缓解率达41.3%,中位生存期也达12.1 月[5]。
TNP-351是由日本武田公司的Miwa和他的同事设计合成的,代表一类新的结构类型的 DHFR抑制剂。采用 2,4-二氨基吡咯并[2,3-d]嘧啶代替叶酸类似物的蝶啶结构,见图4,引入了柔性的三亚甲基桥,提高抑制剂与靶酶活性部位的氢键和疏水键作用。TNP-351、次黄嘌呤和胸苷的组合而不是胸腺嘧啶和 5-氨基-4-甲酰胺咪唑(5-amioimidazole-4-carboxamide,AICA)的组合,被有效地吸收,并通过FPGS在肿瘤细胞中很快转化为聚谷氨酸。 聚谷氨酸不容易从肿瘤细胞中排出,并且可能强烈地抑制 AICARTFase以及DHFR,使得TNP-351对肿瘤有更好的抑制效果[6]。
AAG120-292-3及其类似物为含呋喃环的抑制剂,对不同肿瘤有一定选择性,结构式见图5。呋喃环增强化合物与酶的疏水相互作用并增加亲脂性,这可以提供对肿瘤细胞生长的更有效的抑制,同时经典的脱甲叶酸类似物的桥碳的烷基化可以增强转运到某些肿瘤细胞中的能力,导致了对肿瘤细胞的选择性。生物学结果表明,将 C9-甲基延伸到C8-C9桥区上的乙基使重组人 DHFR的抑制效力翻倍。部分化合物也是胸苷酸合酶的弱抑制剂、FPGS的有效底物,但DHFR为主要的细胞内靶标[7]。
三嗪-苯并咪唑类的化合物 1,2,3均表现出良好的抗肿瘤效果,其分子结构中的苯环与 DHFR形成很强的疏水键,形成了很强的抑酶作用,并且随着疏水性的增加,抑酶作用逐渐增加,其中以1抑制DHFR的作用最为明显,结构式见图6[8]。
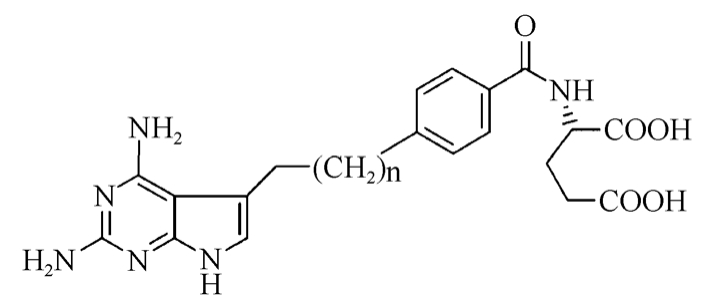
图4 TNP-351
Fig. 4 TNP-351

图5 AAG120-292-3
Fig. 5 AAG120-292-3
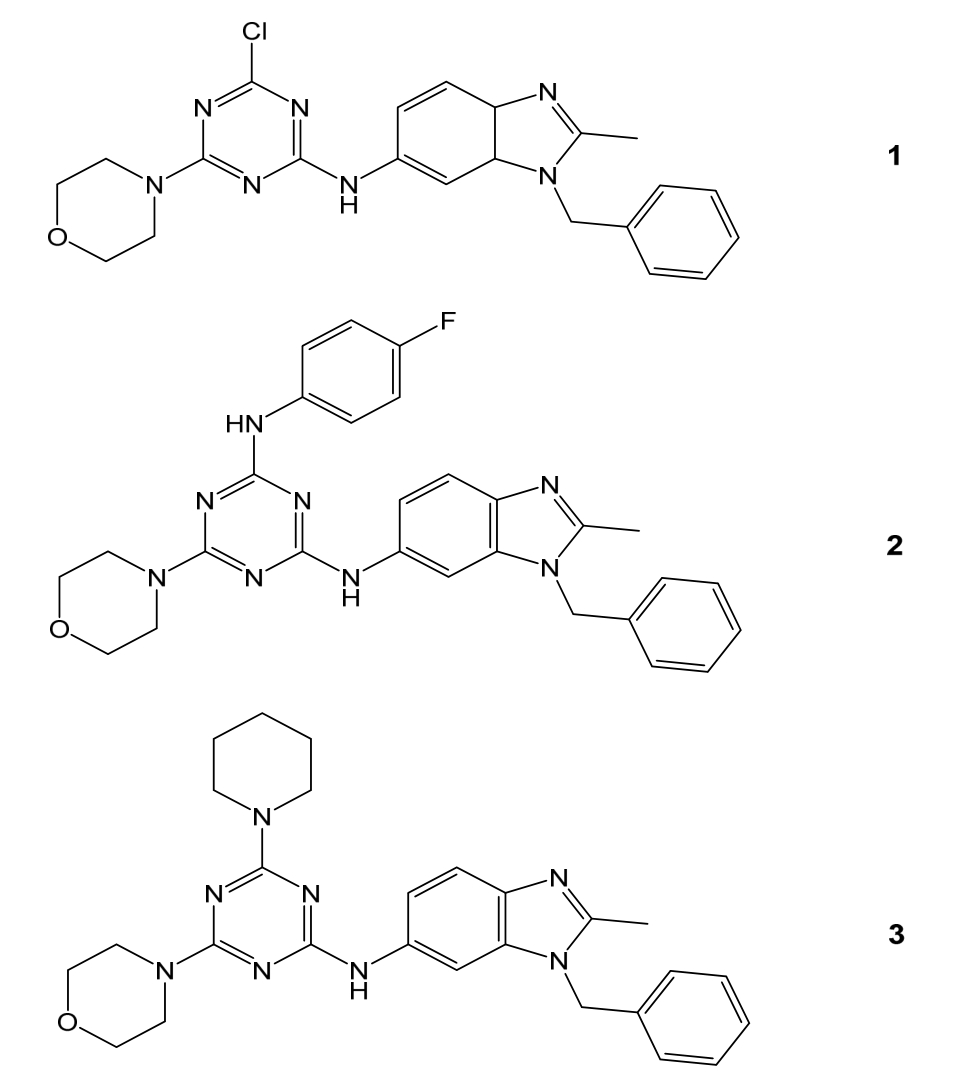
图6 其他经典DHFR抑制剂的结构
Fig. 6 The structure of other classic DHFR inhibitors
非经典 DHFR抑制剂不含谷氨酸残基,脂溶性大,可被动扩散进入细胞.进入细胞不需要多聚谷氨酸化。经典DHFR抑制剂由于是FPGS的底物,往往导致DHFR和FPGS的过分表达,使细胞产生抗药性。而且经典的 DHFR抑制剂在细胞内的滞留时间较长,这导致对正常细胞的毒性较大。相比较而言,非经典 DHFR抑制剂的特点使其有效地降低了耐药性,对正常细胞的毒性也得到了降低,从而提高了药效。代表性的抑制剂包括三甲曲沙、比曲克辛、培美曲塞类似物,结构式见图7。
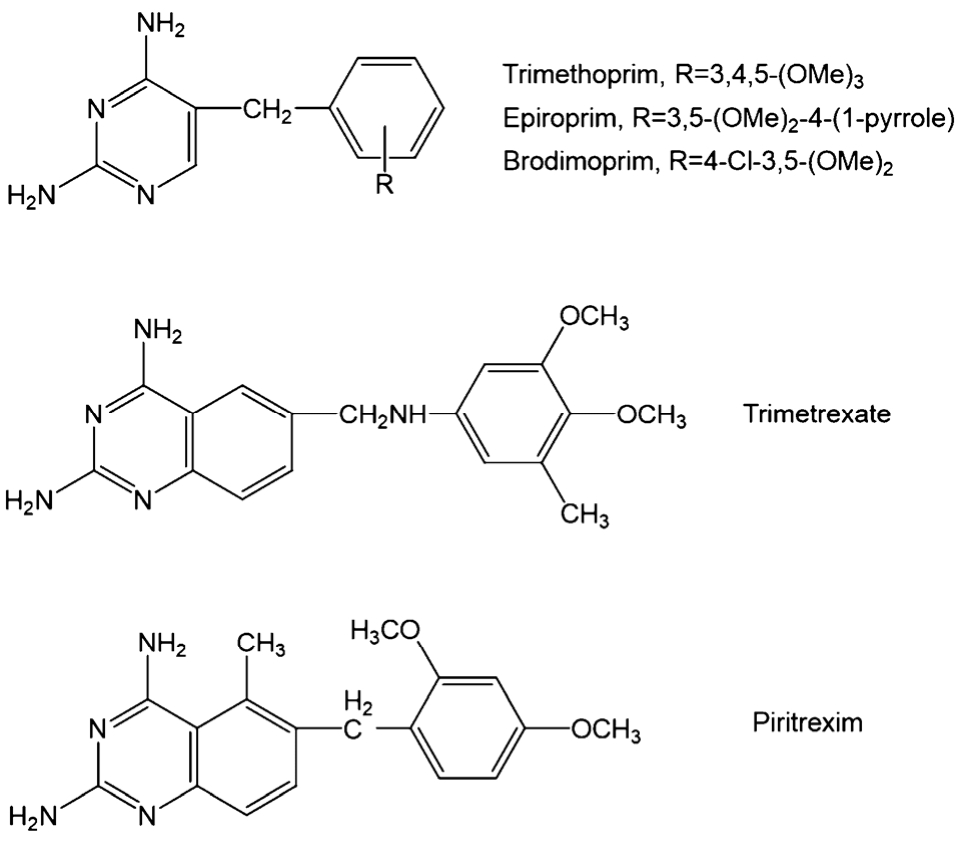
图7 非经典DHFR抑制剂结构
Fig. 7 The structure of nonclassical classic DHFR inhibitors
三甲曲沙(三甲氧苯胺喹唑啉,trimetrexate,TMQ)为亲脂性非经典DHFR抑制剂,为甲氨蝶呤的衍生物,作用机制类似于甲氨蝶呤,不通过RFC转运,它具有更高的脂溶性,能够克服甲氨蝶呤的耐药性。对头颈癌、NSCLC、乳腺癌、胃癌等有明显疗效。它与5-FU及甲酰四氢叶酸合用效果更好[9]。比曲克辛(Piritrexin,PTX)是第 2代非经典DHFR抑制剂,与TMQ作用机制相似,但效力强于TMQ,主要对黑色素瘤、胶质瘤、肉瘤有效,限量毒性主要为骨髓抑制与黏膜炎[10]。甲氧苄氨嘧啶(trimethoprim,TMP)抗菌谱与磺胺药相近,抑制 DHFR,同磺胺药共同作用(磺胺药抑制二氢叶酸合成酶),可使细胞的叶酸代谢受到双重阻滞,提高抗肿瘤作用[11]。依匹普林(epiroprim,EPM)和溴莫普林是TMP类似物,与DHFR具有更强的结合力,而且具有更好的药动学,后者有望代替TMP。
此外,DHFR也是结核病中已知的治疗靶点,甲氨蝶呤作为治疗结核病的有效药物,研究发现其发挥抑制分支杆菌的 DHFR(mt-DHFR)的位点是可能的 DHFR的丙三醇结合位点,基于此,研究者研究开发了4种有效的mt-DHFR新型的抑制剂,也为今后化学合成新的特异性作用于mt-DHFR的靶点提供思路[12]。基于结构和活性的关联性,设计合成了3个吡啶并[2,3-d]嘧啶结构的DHFR抑制剂[13]。
DHFR抑制剂药物抑制肿瘤细胞的 DNA合成,同时对正常增殖细胞的DNA合成也会产生影响。对后者的影响常被认为是抗增殖药物的不良反应,比如胃肠道反应(黏膜炎、腹泻),骨髓毒性(白血球减少症、骨髓抑制)和神经毒性(反应迟缓、共济失调)等。近年来,由于该类抑制剂在幼年和青少年患者中的广泛使用,其神经毒性越来越受到关注,以其经典抑制剂MTX、培美曲塞为例论述其神经毒性。
大剂量 MTX长期用药可导致神经系统的不良反应。研究发现,其神经毒性主要通过作用于星形胶质细胞和神经元发挥毒副作用,大剂量应用MTX和(或)鞘内注射可导致急性、亚急性和慢性神经中毒表现[14]。Weng等[15]研究发现,在小鼠应用MTX之后,其脊髓或视神经中包裹轴突的髓鞘明显减少,并且髓鞘明显变薄,可得知MTX严重损害发育性髓鞘形成,引起中枢神经系统脱髓鞘现象。同时,实验证明MTX不影响少突胶质细胞分化但诱导成熟的少突胶质细胞死亡,并在祖细胞阶段保留大多数少突胶质细胞。但用药后脱髓鞘会再生,不造成永久性损害。
培美曲塞作为新型的 DHFR抑制剂,相较于传统的MTX,其治疗效果更好,而神经毒性反应较轻。
DHFR通过催化二氢叶酸还原为四氢叶酸,从而在DNA的合成和细胞内的叶酸代谢中起着重要的作用,已成为肿瘤化疗的重要作用靶点。DHFR抑制剂药物的结构与其底物相类似,DHFR抑制剂在肿瘤细胞快速增长过程中能选择性地与 DHFR结合,阻止或抑制正常底物与酶的结合,抑制其催化还原活性,使二氢叶酸不能转变成四氢叶酸,阻碍叶酸代谢,干扰DNA和蛋白质的合成,最终导致细胞死亡,从而达到治疗肿瘤的目的,因此,DHFR抑制剂在急性白血病、乳腺癌、绒毛膜上皮癌、骨肿瘤等多种癌症均具有明显的治疗效果。但是随着临床应用扩大,已有的抑制剂药物也表现出了不同程度的不良反应,尤其是神经系统毒性,表现为脑白质病、癫痫、急性蛛网膜炎以及自主神经的损害等,严重影响患者的生活质量,大大限制了其临床应用。同时,现阶段具有神经保护作用的药物如兴奋氨基酸拮抗剂、Ca2+拮抗剂、一氧化氮合酶抑制剂、神经营养因子等,临床应用的效果不明显。综上所述,随着对人体的肿瘤细胞和正常细胞 DHFR的结构差异进一步研究,未来主要研究方向是开发脂溶性大的非经典类 DHFR抑制剂和多靶点抑制剂,同时,明确该类抗肿瘤药物的神经毒性机制,期望能找到选择性高,神经毒性小的DHFR抑制剂。
[1] RJAGOPALAN P T, ZHANG Z, MCCOURT I, et al.Interaction of dihydrofolate reductase with methotrexate:ensemble and singlemolecule kinctics [J]. Proc Natl Acad Sci USA, 2002, 99(21): 13481-13486.
[2] SHARMA V K, ABBAT S, BHARATAM P V. Bharatam.Pharmacoinformatic study on the selective inhibition of the protozoan dihydrofolate reductase enzymes [J]. Molecular Informatics, 2017. Doi: 10.1002/minf.201600156.
[3] HU B B, TANG L N, ZHENG S E, et al. Clinical observation of methotrexate, cisplatin, adriamycin and ifosfamide for stageⅡB extremity osteosarcoma [J]. Chin Clin Oncol(临床肿瘤学杂志), 2013, 18(4): 353-356.
[4] LA MONICA S, MADEDDU D, TISEO M, et al.Combination of Gefitinib and Pemetrexed prevents the acquisition of TKI-resistance in NSCLC cell lines carrying EGFR activating mutation [J]. J Thorac Oncol, 2016, 11(7):1051-1063
[5] SINGLA P, LUXAMI V, PAUL K. Triazine-benzimidazole hybrids: anticancer activity, DNA interaction and dihydrofolate reductase inhibitors [J]. Bioorg Med Chem, 2015,23(8): 1691-700.
[6] MIDDLETON G W, SMITH I E, O’BRIEN M E, et a1. Good symptom relief with palliative MVP chemotherapy in malignant mesothelioma [J]. Ann Oncol, 1998, 9(3): 269-273.
[7] ITOH F, RUSSELLO O, AKIMOTO H, et al. Novel pyrrolo [2,3-d]pyrimidine antifolate TNP-351: cytotoxic effect on methotrexate-resistant CCRF-CEM cells and inhibition of transformylases of de novo purine biosynthesis [J]. Cancer Chemother Pharmacol, 1994, 34(4): 273-279.
[8] SINGLA P, LUXAMI V, PAUL K. Triazine-benzimidazole hybrids: anticancer activity, DNA interaction and dihydrofolate reductase inhibitors [J]. Bioorg Med Chem, 2015,23(8): 1691-700.
[9] DAS J R, FRYAR E B, GREEN S, et al. The protection against trimetrexate cytotoxicity in human bone marrow by sequence-dependent administration of raloxifene,5-fluorouracil/trimetrexate [J]. Anticancer Res, 2006, 26(6B)4279-4286.
[10] MCGUIRE J J. Anticancer antifolates: current status and future directions [J]. Curr Pharm Des, 2003, 9(31): 2593-2613.
[11] ROSOWSKY A, PAPOULIS A T, QUEENER S F. 2,4-Diaminothieno [2, 3-d]pyrimidine lipophilic antifolates as inhibitors of Pneumocystis carinii and Toxoplasma gondii dihydrofolate reductase [J]. J Med Chem, 1997, 40(22):3694-3699.
[12] HONG W, WANG Y, CHANG Z, et al. The identification of novel Mycobacterium tuberculosis DHFR inhibitors and the investigation of their binding preferences by using molecular modelling [J]. Sci Rep, 2015(5): 15328. Doi:10.1038/srep15328.
[13] CODY V, PACE J, NAMJOSHI O A, et al. Structure-activity correlations for three pyrido [2, 3-d]pyrimidine antifolates binding to human and Pneumocystis carinii dihydrofolate reductase [J]. Acta Crystallogr F Struct Biol Commun, 2015,71(Pt 6): 799-803.
[14] LAMBRECHT L, SLEURS C, LABARQUE V, et al. The role of the MTHFR C677T polymorphism in methotrexate-induced toxicity in pediatric osteosarcoma patients [J].Pharmacogenomics, 2017, 18(8): 787-795.
[15] WENG Q J, WANG J J, WANG J Y, et al. Folate metabolism regulates oligodendrocyte survival and differentiation by modulating [J]. Sci Rep, 2017, 7(1): 1-13.
Progress and Study of Dihydrofolate Reductase Inhibitors in Tumor Therapy
LI Dan1, YANG Lijun2, WENG Qinjie2,3*
(1.Center for Drug Evaluation of China Food and Drug Administration,Beijing 100038,China; 2.College of Pharmaceutical Science,Zhejiang University,Hangzhou 310058,China; 3.Center for Drug Safety Evaluation and Research of Zhejiang University,Hangzhou 310058,China)
ABSTRACT:Dihydrofolate reductase(DHFR) is a key enzyme for the synthesis of DNA precursor deoxythymidine(dTMP),which plays an important role in cell proliferation, and is an important target for tumor therapy. Inhibition of DHFR can inhibit dTMP biosynthesis, thereby inhibiting the growth or the appreciation of tumor cells, which shows anti-tumor effect in clinic. In recent years, the neurotoxicity of these drugs has greatly limited its clinical application. Based on this, this paper reviewed the application of DHFR inhibitors in tumor therapy, and introduced the corresponding neurotoxic side effects (drug dose restrictive adverse reaction), to explore the developmentof DHFR inhibitors in tumor therapy and to provide guidance for clinical rational drug use to reduce neurotoxicity.
KEY WORDS:dihydrofolate reductase(DHFR); inhibitor; anti-tumor; neurotoxicity
REFERENCES
中图分类号:R966
文献标志码:A
文章编号:1007-7693(2017)10-1496-05
DOI:10.13748/j.cnki.issn1007-7693.2017.10.031
引用本文:李丹, 杨丽君, 翁勤洁. 二氢叶酸还原酶抑制剂在肿瘤治疗中的研究进展[J]. 中国现代应用药学, 2017, 34(10):1496-1500.
作者简介:李丹,男,硕士,主管药师 Tel: (010)68921694 E-mail: lid@cde.org.cn*
通信作者:翁勤洁,男,博士,副教授 Tel:(0571)88208076 E-mail: wengqinjie@zju.edu.cn
收稿日期:2017-03-20
(本文责编:曹粤锋)