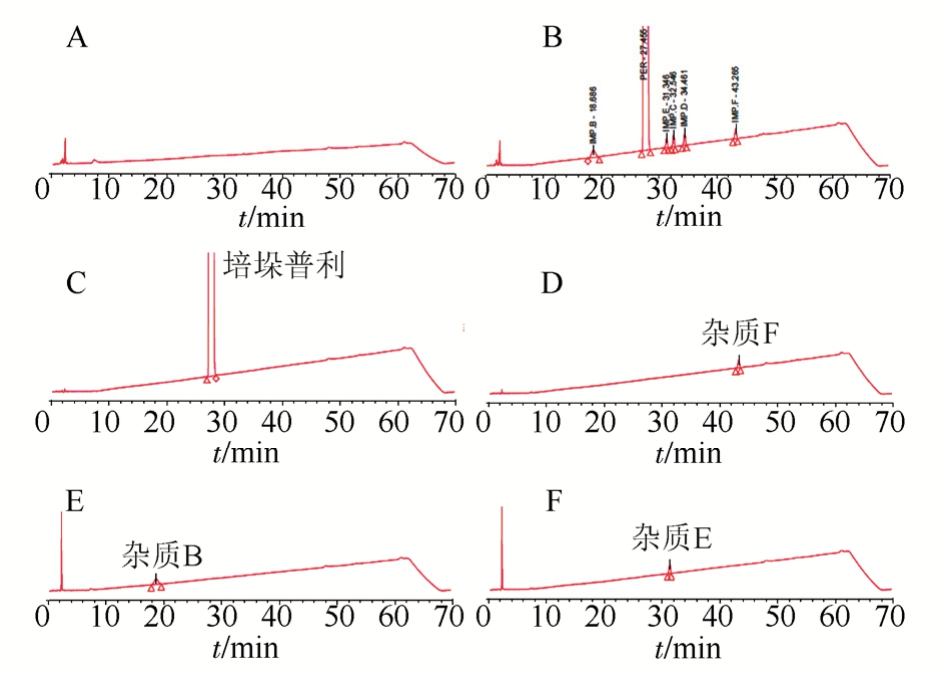
图1 高效液相色谱图
A-空白溶液;B-混合杂质溶液;C-培哚普利对照品溶液;D-杂质F溶液;E-杂质B溶液;F-杂质E溶液。
Fig. 1 HPLC chromatograms
A-blank solution; B-mixed impurity solution; C-perindopril standard solution; D-impurity F solution; E-impurity B solution; F-impurity E solution.
余永华1,马敏康2,杨仲杰1,石晓宝1,张婷1
(1.杭州新诺华医药有限公司,杭州 310015;2.浙江省药品化妆品审评中心,杭州 310012)
摘要:目的 采用加校正因子的主成分自身对照法测定培哚普利片中有关物质的含量。方法 采用GL Inertsil C8-3色谱柱(4.0 mm×150 mm,5 μm),以流动相A(用50%高氯酸调节pH至2.5的水溶液)和流动相B(0.03%的高氯酸乙腈溶液)进行梯度洗脱,流速为1.0 mL·min-1,检测波长为215 nm,进样量为20 μL,柱温60 ℃,测定培哚普利叔丁胺和杂质B、E、F的线性方程,以斜率计算杂质相对于培哚普利叔丁胺的校正因子,用相对保留时间确定各杂质位置。并进行了方法学验证。用相对校正因子计算培哚普利片中杂质B、E和F的含量,并与杂质对照品法测得的结果进行比较。结果 培哚普利杂质B、E和F的相对保留时间分别为0.68,1.14,1.61;校正因子分别为0.77,1.02,1.24;检出限分别为2.24,0.52,1.02 ng;定量限为6.73,1.57,3.06 ng;采用校正因子的主成分自身对照法和外标法测得的结果无显著性差异。结论 该方法简便快速,可准确测定培哚普利片中杂质B、E和F含量。
关键词:培哚普利片;有关物质;校正因子
培哚普利是一种使血管紧张素 I转化为血管紧张素II的酶(血管紧张素转化酶)抑制剂,临床上用于治疗高血压和充血性心衰[1]。中国药典 2015年版已收载培哚普利叔丁胺及培哚普利叔丁胺片[2],其有关物质检查采用 HPLC,但仅对杂质IV(即杂质E)、单杂和总杂进行控制。经对比BP[3]、USP[4]中的培哚普利叔丁胺及片剂质量标准,发现国外药典对有关物质的控制更加严格,尤其在杂质个数上,如对杂质B、C、D、E、F均进行了严格控制。
根据文献研究[5-7]表明培哚普利叔丁胺在高温及高湿等条件下分别水解或环合产生杂质 B和杂质E,杂质F在水溶液下经高温及酸破坏产生杂质C和杂质D。因此杂质B和杂质F为培哚普利的一级降解杂质,是产品质量控制的关键,E为工艺过程中的杂质。其中,杂质B和F的结构式与培哚普利叔丁胺存在明显的差异,因此其响应因子可能存在较大差异,而查阅有关培哚普利药典标准及质量研究的相关文献[8],尚无采用校正因子法对培哚普利叔丁胺片中杂质B、E、F进行控制的报道。
综上所述,当前培哚普利片的有关物质研究在已知杂质的控制上存在不足,而目前对单个杂质的 HPLC测定方法主要有内标法、外标法、面积归一化法、加校正因子的主成分自身对照法和不加校正因子的主成分自身对照法[9-11]。鉴于目前培哚普利叔丁胺的杂质对照品较难获得,且不加校正因子的主成分自身对照法在杂质限量方面存在的明显缺陷,本研究对培哚普利的有关物质B、E和F的校正因子进行研究,并建立加校正因子的主成分自身对照法测定培哚普利叔丁胺片中有关物质含量的方法。
1.1 仪器
Waters e2695/2498/2998高效液相色谱仪(美国Waters公司);XS 205DU电子分析天平、AL-204电子分析天平和FE20pH计均购自瑞士梅特勒公司。
1.2 试药
乙腈(色谱纯,TEDIA);高氯酸(分析纯,天津鑫源化工);水为纯化水;培哚普利叔丁胺盐(中国食品药品检定研究院,批号:101072-201101,纯度:99.2%),杂质 B(USP,批号:F0L113、纯度:95.0%),杂质F(USP,批号:F0L129,纯度:91.0%),培哚普利峰鉴别对照品(EP,批号:1.3),杂质C(USP,批号:F025Q0,纯度:100.0%),杂质D(USP,批号:F0L144,纯度:100.0%),杂质E(TLC,批号:1836-002A3,纯度:99.4%)。培哚普利片[施维雅(天津)制药有限公司,批号:2007410]。
2.1 溶液的制备
2.1.1 对照品溶液制备 储备溶液的制备:取培哚普利叔丁胺,杂质B、E、F对照品适量,精密称定,加流动相A使溶解并定容,摇匀,分别配制成浓度为 172.61,74.86,78.33,76.44 μg·mL-1的对照品储备溶液,备用。
2.1.2 系统适应性溶液的配制 峰鉴别溶液:取培垛普利峰鉴别对照品(含杂质 B、E、F、H、K)适量,加流动相A使溶解并制成每1 mL中约含1.6 mg的溶液,备用。杂质C和杂质D混合溶液:分别取杂质C和杂质D适量,加流动相A使溶解并制成含杂质C和杂质D每1 mL中各含0.1 mg的溶液,备用。
2.1.3 供试品溶液及空白辅料溶液制备 取本品细粉适量(约相当于培哚普利叔丁胺盐16 mg),置10 mL量瓶中,加流动相A适量,超声使培哚普利叔丁胺溶解,用流动相A稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液。
空白辅料溶液:取处方量的空白辅料(不含培哚普利),置10 mL量瓶中同供试品溶液操作。
2.1.4 对照溶液及灵敏度溶液的制备 精密量取供试品溶液1 mL,置200 mL量瓶中,用流动相A稀释至刻度,摇匀,作为对照溶液;精密量取对照溶液1 mL,置10 mL量瓶中,用流动相A稀释至刻度,摇匀,作为灵敏度溶液。
2.2 色谱条件及系统适用性
色谱柱为 Inertsil C8-3(4.0 mm×150 mm,5 μm),以流动相 A(用 50%高氯酸调节 pH 至 2.5的水溶液)和流动相B(0.03%的高氯酸乙腈溶液)进行梯度洗脱:0~5 min,95%A;5~60 min,95%→40%A;60~65 min,40%→95%A;65~75min,95%A,流速为1.0 mL·min-1,检测波长为215 nm,进样量为20 μL,柱温60 ℃。
系统适用性:培哚普利峰保留时间约为25 min,杂质B、K、E、F、H相当于培哚普利叔丁胺的保留时间分别为 0.68,0.72,1.14,1.61,1.79,峰谷比Hp/Hv≥3(Hp为杂质B到基线的高度,Hv为杂质B和K交点的最低点到基线的高度)。
2.3 方法学验证[12-13]
2.3.1 系统适用性 取系统适用性溶液和灵敏度溶液按“2.2”项色谱条件连续进样6针,记录相对保留时间,峰面积,峰高比,主峰拖尾因子,灵敏度溶液信噪比。
6针图谱中,各杂质B、K、E、F、H相对保留时间RSD均<1.0%,各杂质峰面积RSD<2.0%,峰高比均>3,分离度均>3。灵敏度溶液信噪比均>10。
2.3.2 专属性
2.3.2.1 已知杂质及空白干扰 分别取空白辅料溶液、杂质B、杂质E、杂质F、混合杂质溶液(含杂质B、C、D、E、F)、培哚普利对照品溶液,考察空白干扰及已知杂质及主峰之间的分离度和峰纯度。
溶剂和空白辅料对杂质和主峰均无干扰,各杂质之间及与主峰之间分离度均无干扰,分离度均>1.5。结果见图1。
图1 高效液相色谱图
A-空白溶液;B-混合杂质溶液;C-培哚普利对照品溶液;D-杂质F溶液;E-杂质B溶液;F-杂质E溶液。
Fig. 1 HPLC chromatograms
A-blank solution; B-mixed impurity solution; C-perindopril standard solution; D-impurity F solution; E-impurity B solution; F-impurity E solution.
2.3.2.2 破坏实验 酸、碱、高温、光照破坏实验:称取样品细粉适量(约相当于培哚普利 16 mg)5份,分别置10 mL量瓶中,分别进行酸、碱、高温、光照破坏后,加流动相稀释至刻度,过滤,取续滤液即得酸、碱、高温、光照破坏性溶液。破坏性操作如下:①酸破坏,加6 mol·L-1的盐酸溶液2 mL,室温静置4 h,用6 mol·L-1的氢氧化钠溶液2 mL中和;②碱破坏,加0.2 mol·L-1氢氧化钠溶液2 mL,室温静置1 h,用0.2 mol·L-1的盐酸溶液2 mL中和;③高温破坏,样品加流动相A 2 mL,超声使培哚普利溶解,置80 ℃水浴加热2 h,放冷;④光照破坏,样品加流动相A 2 mL,超声使培哚普利溶解,至4 500 lx光照条件下光照7 d;⑤氧化破坏,样品加入30%的过氧化氢溶液2 mL,超声使培哚普利溶解,室温放置3 h。
取各破坏溶液,按“2.2”项下色谱条件进样,结果显示本品在光照条件下相对稳定,产生少量的杂质B;酸、碱条件下主要降解杂质为杂质B;氧化条件下主要降解成杂质 F及其他未知杂质;而高温条件下除降解产生杂质 B外,也有杂质 F产生。在“2.2”项色谱条件下,主要降解杂质均能有效分离。结果显示本法专属性良好。结果见图2。

图2 高效液相色谱图
A-酸破坏;B-碱破坏;C-氧化破坏;D-光照破坏;E-高温破坏。
Fig. 2 HPLC chromatograms
A-acid destruction; B-base destruction; C-ioxidation destruction;D-light destruction; E-heat destruction.
2.3.3 检测限与定量限 取培哚普利、杂质 B、杂质E、杂质F混合溶液,逐级稀释,使各杂质主峰信噪比约为3︰1和10︰1,记录对应的对照品溶液浓度,即为对应的检测限与定量限,按“2.2”项下色谱条件进样检测。
结果,培哚普利、杂质B、杂质E、杂质F的定量限分别为0.35 ng(RSD=1.1%,n=6),2.24 ng(RSD=0.8%,n=6),0.52 ng(RSD=1.1%,n=6),1.02 ng(RSD=1.4%,n=6),检测限分别为 1.04,6.73,1.57,3.06 ng。
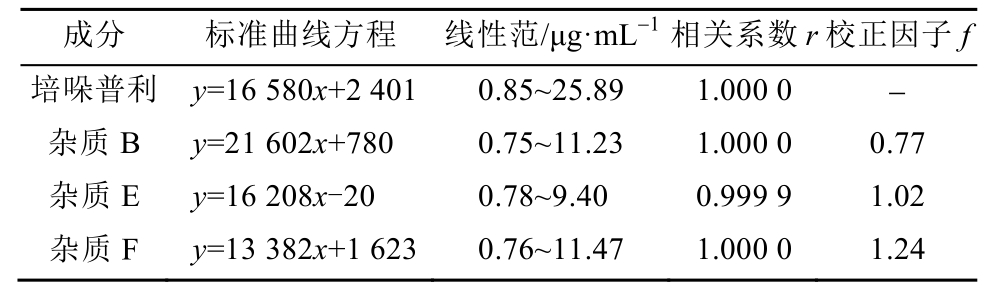
2.3.4 线性及校正因子 精密量取培哚普利对照品储备液 0.05,0.25,0.4,0.8,1.0,1.5 mL和杂质(B、F)储备液 0.1,0.5,0.8,1.0,1.2,1.5 mL及杂质E储备液0.1,0.4,0.6,0.8,1.0,1.2 mL置10 mL量瓶中,用流动相A稀释使各杂质浓度分别为0.05%至150%限度范围内不同浓度的溶液(杂质B和杂质F限度0.5%,杂质E限度0.4%);按“2.2”项色谱条件进样检测,并以峰面积(y:纵坐标)对浓度(x:横坐标)进行线性回归,得线性方程。校正因子计算公式为f=r培哚普利/r杂质(r为线性方程斜率)。
结果表明,培哚普利叔丁胺、杂质 B、E、F的线性范围良好,相关系数分别为 1.000 0,1.000 0,0.999 9,1.000 0。杂质B、E和F的校正因子分别为0.77,1.02,1.24。结果见表1。
2.3.5 回收率 取自研产品(宁波美诺华天康药业有限公司,批号:150901)细粉适量(约相当于培哚普利叔丁胺盐16 mg,其中杂质B、E、F含量分别为0.02%,0.00%,0.05%),置10 mL量瓶中,分别加入杂质B、E、F对照品储备溶液适量,使成含杂质B、E和F分别为定量限、限度浓度(80%,100%,120%)的供试品溶,各浓度平行制备3份。按“2.2”项下色谱条件进样检测,记录峰面积(Y),并计算回收率和 RSD[回收率计算公式:回收率(%)=测的量-已知量/加入量×100%。
表1 培哚普利及3种杂质的标准曲线方程、线性范围、相关系数和校正因子
Tab. 1 Regression equations, linear ranges, correlation coefficient, relative correction factors of peridopril and two impurities
结果表明,杂质B、E、F的回收率良好。杂质B各浓度下回收率(n=3)分别为101.8%,102.3%,102.7%;总回收率(n=9)和 RSD分别为 102.2%和0.7%。杂质E各浓度下回收率(n=3)分别为98.4%,99.3%,100.2%;总回收率(n=9)和 RSD 分别为99.3%和1.0%。杂质F各浓度下回收率(n=3)分别为98.9%,98.4%,99.0%;总回收率(n=9)和RSD分别为98.8%和0.6%。
2.3.6 精密度 按“2.1.3”项下方法,平行配制6份供试品溶液,按“2.2”项下色谱条件进样检测,记录峰面积(Y),计算杂质 B、E和杂质 F的含量及相对标准偏差(RSD),作为重复性。另以不同人员同法操作,以2位操作者12针样品的峰面积,计算12次的相对标准偏差,作为中间精密度。
结果,本法精密度良好,杂质B重复性RSD为0.6%,中间精密度RSD为1.1%。杂质E重复性RSD为0.5%,中间精密度RSD为1.2%。杂质F重复性RSD为0.6%,中间精密度RSD为0.6%。
2.3.7 耐用性 考察流速变化±20%(0.8,1.2 mL·min-1)、柱温变化±5 ℃(55,65℃)、pH值变化±0.2(2.3,2.7)、不同批号色谱柱条件下对系统适用性、供试品检测结果影响,结果表明流速、柱温、pH值、色谱柱变化后,对系统适用性和供试品检测结果无影响,表明方法耐用性良好。
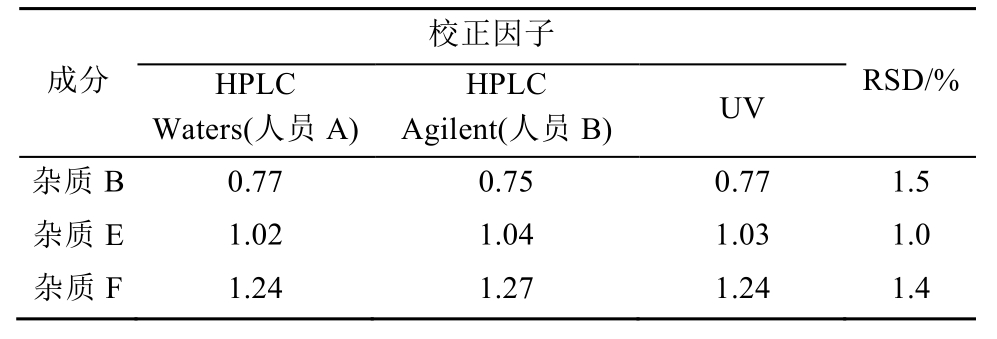
校正因子的耐用性:采用“2.3.4”项下的溶液配制方法,考察不同的液相(Waters、Agilent)、不同的人(人员A和B)、不同方法(UV单点法、线性法)在不同时间下检测对校正因子的影响。结果显示,杂质B、杂质E、杂质F校正因子耐用性良好(RSD<2%)。结果见表2。
表2 培哚普利及2种杂质的校正因子耐用性
Tab. 2 Relative correction factors durability of peridopril and two impurities
2.3.8 滤膜吸附性 取“2.3.4”项下的对照品混合溶液分别过滤0,2,4,6,8 mL的续滤液,按“2.2”项下色谱条件进样检测,记录峰面积(Y),并以过滤0 mL溶液为100%浓度计算各续滤液的回收率。结果杂质B、E和杂质F均无明显滤膜吸附,各续滤液的回收率均>98%。
2.3.9 溶液稳定性 取“2.3.4”项下对照品溶液和“2.3.6”项下供试品溶液,分别测定在常温和5 ℃条件下0,4,12,24 h的稳定性,按“2.2”项下色谱条件进样,记录色谱峰面积,计算有关物质含量。结果,常温下供试品溶液不稳定,24 h杂质B含量增加>2%;而在5 ℃,24 h杂质B、E、杂质F对照品溶液变化<2.0%,供试品溶液中杂质含量变化<2.0%,且无未知杂质产生。
2.4 样品测定
取培哚普利片3批,按“2.1”项下供试品、对照溶液、对照品溶液的配制方法,按“2.2”项下色谱条件进样,进行含量测定。分别按加校正因子的主成分自身对照法和外标法计算有关物质的含量,结果2种方法对供试品中杂质B、杂质E、杂质F的含量计算结果相似(差值<0.05%)。结果见表3。
3.1 波长的选择
对培哚普利叔丁胺、杂质B、杂质E和杂质F进行波长扫描,结果最大吸收波长分别为 215,213,205,207 nm,由于培哚普利叔丁胺为短波长吸收,为兼顾三者的检测效能,综合考虑选择215 nm作为检测波长。
3.2 供试品溶液的稳定性
本实验研究表明培哚普利叔丁胺片供试品溶液虽然在进样过程中有关物质无明显增加,但其溶液放置稳定性欠佳,此点并未在BP及中国药典标准收载,供试品溶液应在低温下保存或临用新配。
表3 有关物质检测结果
Tab. 3 The determination result of related substances %
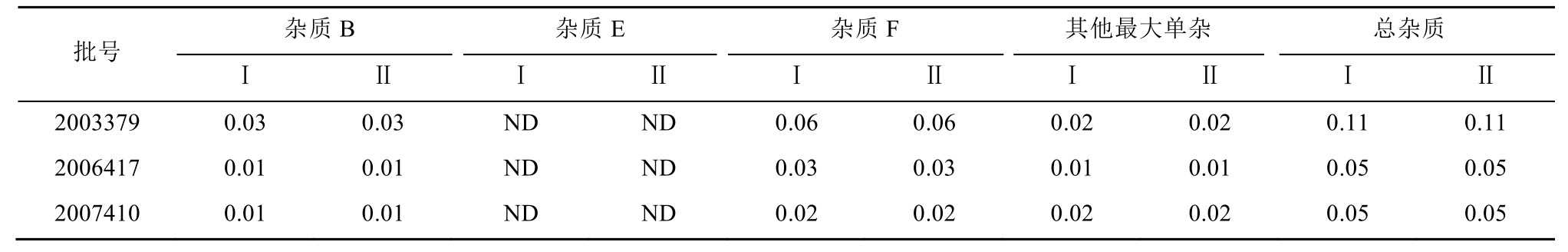
注:Ⅰ为外标法,Ⅱ为加校正因子的主成分自身对照法。
Note: Ⅰ was external standard method, Ⅱ was the main component self-compare with calibration factor method.
3.3 校正因子法
本实验以培哚普利叔丁胺为参照物建立校正因子法,阐明了培哚普利叔丁胺与 2个降解杂质间的相互关系,并考察了不同的实验条件下对校正因子测定的影响。结果采用外标法与校正因子法测定的结果无显著性差异,表明本法测定的校正因子准确性好,可作为培哚普利叔丁胺片中杂质B和杂质F的检测方法。培哚普利叔丁胺杂质B、E、F的相对保留时间分别0.68,1.14,1.61,校正因子分别为0.77,1.02,1.24,其中杂质E的校正因子在0.8~1.2内,实际应用时修约为1[14],不予校正。
3.4 小结
本研究根据中国药典中培哚普利叔丁胺片标准,在此基础上增加了对已知降解杂质的校正因子进行了研究,经方法学研究表明,本方法简便快速,可准确测定培哚普利片中降解杂质B、E和F含量,且灵敏可用,对培哚普利的质量控制具有重要意义。
[1] TODD P A, FITTON A. Perindopril. A review of its pharmacological properties and therapeutic use in cardiovascular disorders [J]. Drugs, 199, 42(1): 90-114.
[2] 中国药典. 二部[S]. 2015: 1201-1202.
[3] Perindopril Erbumine Tablets. British Pharmacopoeia [S].2015, Ⅲ-968.
[4] United States Pharmacopoeia 39-NF34 [S]. 2016: 8130.
[5] COVERSYL®Tablets. 日本標準商品分類番号872144(2014年6月改訂(第19版): 5.
[6] YAO C Z, YU Y H, SHI X B. A stable alpha crystal form of perindopril t-butyl amine tablet and a preparation method thereof: CN, 201510885002. 8 [P]. 2015-12-04.
[7] HUANG L Q, BIAN C A. A perindopril tablet and its powder direct tabletting process: CN, 201510040697.X [P].2015-01-07.
[8] SZABÓ Z I, RÉTI Z Z, GAGYI L, et al. Simultaneous quantification of related substances of perindopril tert-butylamine using a novel stability indicating liquid chromatographic method [J]. J Chromatogr Sci, 2015, 53(3):424-430.
[9] 中国药典. 四部[S]. 2015: 附录0512.
[10] XIE M F. How to establish an HPLC method for determination of related substances in drugs [J]. Chin J Pharm(中国医药工业杂志), 2007, 38(1): 45-48.
[11] HU L N, ZHANG Y, GU J, et al. Determination of the content of impurity D in atorvastatin calcium with the corretion factor[J]. Chin J Mod Appl Pharm(中国现代应用药学), 2015,32(12): 1476-1480.
[12] International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. Validation Of Analytical Procedures: Methodology Q2B.1994: 2-8.
[13] 中国药典. 四部[S]. 2015: 附录9101.
[14] British Pharmacopoiea. Volume V-Appendices-Appendix III Chromatographic Separation Techniques [S]. 2015: Ⅴ-188.
Main Component Self-compare with Calibration Factor Method for Determination of Related Substances in Perindopril Tablets
YU Yonghua1, MA Minkang2, YANG Zhongjie1, SHI Xiaobao1, ZHANG Ting1
(1.Hangzhou SINOVO Pharmaceutical Co., Ltd., Hangzhou 310015, China; 2.Zhejiang Center for Drug & Cosmetic Evaluation, Hangzhou 310012,China)
ABSTRACT:OBJECTIVE To estabilish the main component self-compare with calibration factor method for determination of related substances in perindopril tablets. METHODS The GL Inertsil C8-3 column (4.0 mm×150 mm, 5 μm) was adopted with the mobile phase composed of water (adjusted to pH 2.5 with 50% perchloric acid) and acetonitrile solution(contain with 0.03% perchloric acid), pumped at a flow rate of 1.0 mL·min-1, the detection was carried out at 215 nm and the column temperature was 60 ℃ , the injection volume was 20 μL. The slope of linear equation was used to determine the correction factor between impurity B, E, F and perindopril. The relative retention on time was used to determine the position of impurities. The developed method was validated. The results obtained by the proposed method were compared with those by using the reference standards of impurities to verify the method accuracy. RESULTS The relative retention time of imputiry B, E, F was 0.68, 1.14,1.61, respectively. The correction factor of imputiry B, E, F was 0.77, 1.02, 1.24, respectively. The limits of detection and quantification of imputiry B, E, F were found to be 2.24, 0.52, 1.02 ng and 6.73, 1.57, 3.06 ng, respectively. There were no significant diferences in the results determined by the external and by the self-compare with calibration factor method.CONCLUSION The method is simple, efficient and accurate for analyzing the related substances B, E and F in perindopril tablets.
KEY WORDS:perindopril tablets; related substances; correction factor
REFERENCES
中图分类号:R917
文献标志码:B
文章编号:1007-7693(2017)10-1427-05
DOI:10.13748/j.cnki.issn1007-7693.2017.10.014
引用本文:余永华, 马敏康, 杨仲杰, 等. 加校正因子的主成分自身对照法测定培哚普利片中有关物质的含量[J]. 中国现代应用药学, 2017, 34(10): 1427-1431.
作者简介:余永华,男,硕士,工程师 Tel: (0571)88073919 E-mail: yhua85@sina.com
收稿日期:2017-02-18
(本文责编:李艳芳)