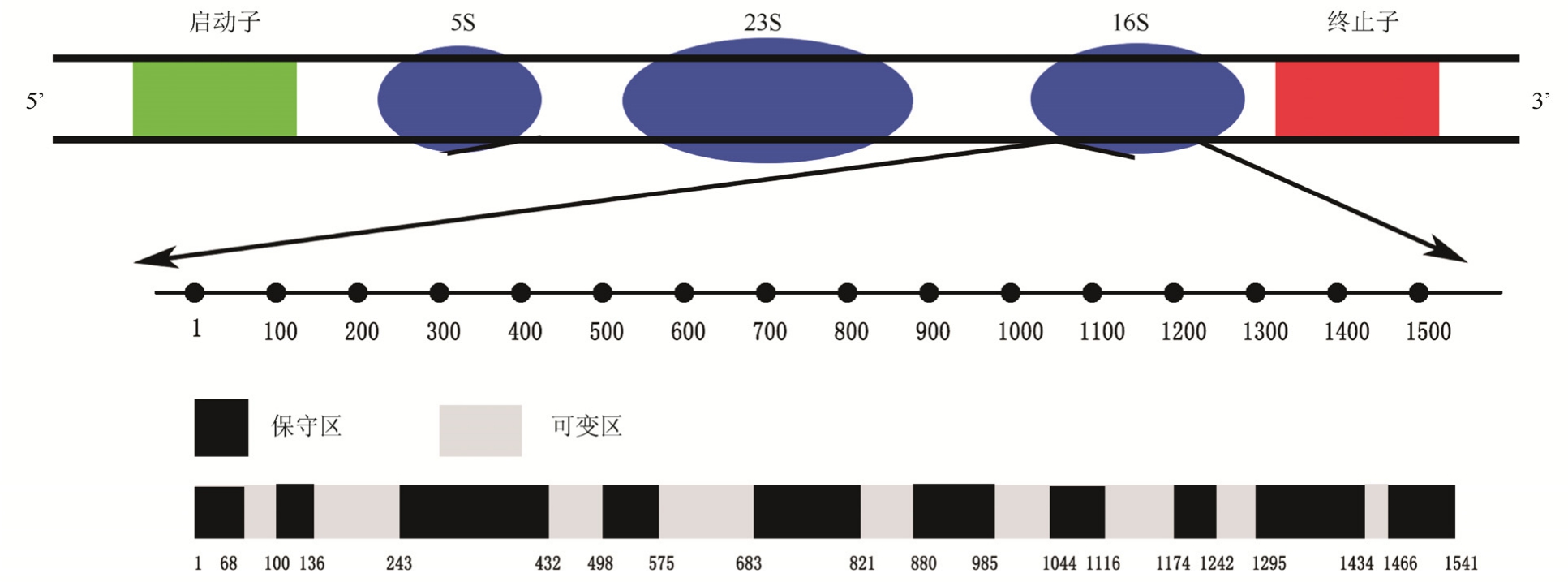
图1 rRNA结构及16S rRNA的保守区与可变区
Fig. 1 The structure of rRNA and the variable region and conserved region of 16S rRNA
·综 述·
张国林1,2,苏远科3,沈海英1,顾珉1,邢以文1
(1.苏州市药品检验检测研究中心,江苏 苏州 215000;2.南京中医药大学,南京 210000;3.龙口出入境检验检疫局,山东 龙口 265700)
摘要:经典的微生物鉴定有赖于微生物的纯培养,其在一些难培养、生长缓慢或需要特殊营养的微生物鉴定方面有明显的局限性。全自动细菌鉴定及药敏分析系统和基质辅助激光解离飞行时间质谱对微生物鉴定属表型鉴定,严重依赖仪器生产厂商提供的数据库,对类型多样的环境微生物的鉴定分型有明显缺陷。基于遗传物质的分子生物学鉴定技术为微生物的准确鉴定及分型提供了新的思路。16S rRNA/ITS遗传信息具有相对稳定性和易变异的双重特点,不依赖于微生物的营养及生长状态,在微生物的鉴定、分型中得到广泛应用。微生物是影响药品质量的重要因素,而药品微生物控制是药品质量控制的重要方面。本文对16S rRNA/ITS序列与微生物鉴定、分型及其在药品微生物质量控制领域中的应用进行综述,为相关工作提供参考。
关键词:微生物鉴定;溯源分析;形态学鉴定;16S rRNA;16S-23S rRNA基因间隔区;分子生物学技术
微生物的快速鉴定分型是微生物学研究的重要内容,也是药品质量控制的重要方面。目前微生物的鉴定分型主要采用形态学、生化反应、免疫学及分子生物学手段进行。经典的鉴定方法有赖于微生物的纯培养,存在耗时长、敏感度低和特异性较差的缺点,在一些难培养(极端微生物)、生长缓慢(如分支杆菌要 4~8周才能长成典型菌落)、代谢不典型或需要特殊营养的微生物鉴定方面有明显的局限性,难以满足实际工作的需要。全自动细菌鉴定及药敏系统属表型鉴定范畴,受鉴定卡内生化反应底物浓度、培养基营养成分的影响校大。同时,细菌本身某些生化反应不典型、菌体受到损伤、药品生产过程干扰微生物的代谢也影响鉴定准确性。全自动细菌鉴定及药敏系统数据库的大小及更新速度也是影响鉴定结果的重要因素。基于非培养基础的分子生物学鉴定分型方法可以有效绕开微生物的纯培养过程,具有快速、准确、高效的优点。16S rRNA(16S Subunit Ribosomal RNA)基因序列具有普遍存在性和序列保守性的特点,是从遗传进化角度和分子水平进行细菌的分类鉴定的物质基础。已有的证据显示,基于关键基因序列信息分析的系统分类结果与基于全基因组信息的分析结果相似[1],且目前已经有基于关键区段分析鉴定微生物的商业化产品应用。目前,16S rRNA已广泛应用于细菌分类、鉴定领域,且随着高通量测序技术(high-throughput sequencing,HTS)、大数据分析方法的应用和致病菌基因序列的不断测序分析,基于16S rRNA序列分析已经成为现代微生物鉴定、分类及溯源研究的主要手段[2-3]。内部转录间隔区(internal transcribed space,ITS)是核糖体大小亚基基因之间的间隔序列,其在进化中的选择压力更小,具有更大的变异性。16S rRNA与ITS序列的结合分析对不同种属间和亲缘关系接近的微生物鉴定方面具有更大的优越性。
微生物控制是药品生产洁净环境控制的重要内容,也是药品质量控制的关键方面,而微生物的快速、准确鉴定,进而实现溯源是微生物控制效果的最关键点之一。本文对16S rRNA/ITS的基因序列在药品微生物控制领域的应用进行综述,为相关工作提供参考。
rRNA的核苷酸序列、二级结构和功能具有进化的保守性和种属的特异性,是研究微生物系统进化和分类最常用的分子标记。5S rRNA序列较短(120 bp),缺乏足够的遗传信息,23S rRNA序列相对较长(2 904 bp),不易分析。而16S rRNA是细菌内含量最大的遗传物质,长度适中(1 541 bp),存在保守区和可变区,是微生物进行鉴定和分型的最好标志物,见图1。
不同种属微生物的 16S rRNA基因序列一级结构差较大,但二级结构在进化中高度保守。核酸的二级结构是rRNA功能维系的重要基础。Pei等研究发现Thermoanaerobacter tengcongensis基因16S rRNA在一级结构方面有6.7%的差异,而在二级结构上却高度保守[4]。因此,16S rRNA在维持功能结构基础上的一级结构变异可以通过自然选择。
内部转录间隔区是核糖体大小亚基基因之间的间隔序列[5]。由于ITS序列面临较小的进化选择压力,在进化过程中比rRNA产生更多的变异性。ITS序列在亲缘关系较近的种属分型方面具有更广泛的应用[6]。Sulaiman等通过对rRNA编码区域内的ITS1进行PCR扩增产物测序分析了希腊酸奶中分离的霉菌类型[7]。在内部转录间隔区内通常含有一些功能基因单位,如tRNA基因等[8]。内部转录间隔区序列的分析可作为微生物鉴定的重要依据。ITS序列中的功能片段和识别序列只占很小的比例,而大部分序列都是以引起变异的无义序列的形式存在。早期的研究证实 ITS片段的进化速率是16S rRNA进化速率的10倍[9]。因此,在不同条件下存在的同种微生物 ITS序列通常有明显的差异性,这为无菌药品检测实验室检出菌的溯源分析提供了新思路。某些微生物之间的16S rRNA序列几乎相同而DNA的相似度却低于70%,表明具有相同16S rRNA序列的微生物却是不同的种。Sashida等通过分析Mycoplasma haemomuris的16S-23S rRNA内部转录间隔区序列将其分成了2个不同的基因进化簇[6]。
图1 rRNA结构及16S rRNA的保守区与可变区
Fig. 1 The structure of rRNA and the variable region and conserved region of 16S rRNA
16S-23S rRNA及23S-5S rRNA之间的序列通常称为ITS,而ITS序列内部编码tRNA基因序列及酶切识别位点序列以外的非保守区段称为inter-genic spacer region,即ISR。ITS序列的差异性主要体现在不同的层次上:不同细菌间 ITS所隶属的操纵子的拷贝数不同;不同种间的 ITS片段长度的差异性以及同一种内不同操纵子间 ITS序列和长度的差异性。ITS序列分析主要应用于细菌的种或亚种的细致分类和细菌种群的结构变化[10-12]。由于 ITS序列的高度可变弥补了 16S rRNA序列的保守性强、变异程度弱的缺点,是细菌的种和亚种水平鉴定的有力工具。ITS序列的高度可变性使其应用受到一定程度的限制,但随着ITS序列数据库的不断扩容和分析方法的改进,其在细菌分类鉴定和种群分析中的作用将越来越重要。
16S rDNA是细菌染色体上编码16S rRNA的DNA序列,由可变区和保守区交叉排列构成。其中保守区在各种细菌中具有种属的稳定性,而可变区具有遗传的变异性,其变异程度与细菌的系统发育过程相关。根据基因序列的相似程度判定微生物种属是微生物鉴定的重要手段,Bosshard等[13]将16S rRNA序列同源性>99%的定义为同种细菌,同源性介于95%~99%的界定为同属的细菌。内部转录间隔区序列是位于 rRNA基因之间的片段,其进化效率要明显高于16S rRNA序列,具有更高的变异性和更多的变异区,是进行微生物鉴定的必要补充。16S-23S rRNA间隔序列可用于不同菌种间的鉴别,还可用于16S rRNA基因序列无法鉴定的非常接近的种间鉴别以及种内不同菌株的鉴别。笔者认为对微生物进行16S rRNA/ITS序列分析鉴定、分类微生物时应优先依据16S rRNA进行初步分类,当涉及到种和亚种及菌株的鉴定或溯源分析时,应着重考虑 ITS序列。目前常用的用于扩增rRNA的引物序列见表1。
分子生物学技术在微生物鉴定及分型方面已有广泛的应用,如免疫杂交、RFLP、DGGE、FISH、SSCP和分子杂交等[14]。16S rRNA/ITS结合HTS为微生物的鉴定、分类及溯源提供了强有力的技术保障。
表1 常用的16S rRNA序列PCR反应扩增引物
Tab. 1 The frequently used primers for PCR of 16S rRNA

2.1 16S rRNA/ITS和荧光原位杂交技术与微生物鉴定
荧光原位杂交(fluorescence in situ hybridization,FISH)是基于放射性原位杂交技术的非放射性分子细胞遗传技术,即采用荧光标记取代同位素标记的原位杂交方法。荧光原位杂交的基本原理是将核酸探针用特定的核苷酸进行标记,然后将探针直接杂交到靶染色体或DNA上,再用与荧光素分子偶联的单克隆抗体与探针分子特异性结合来实现对DNA序列的定性、定位或相对定量分析。Petrich等[15]以梅毒螺旋体16S rRNA为靶序列设计了FISH探针TPALL,将其固定于硅胶上,实现了对组织样本梅毒螺旋体的原位杂交检测。Karlsson等通过使用3类16S rRNA寡聚核苷酸探针,采用荧光原位杂交和HTS实现了对密螺旋体属微生物的鉴定[16]。三氯生是环境中最常见的污染物,但对能降解三氯生的微生物研究甚少。研究人员通过分析来源于(13)C标记的三氯生富集微生物的16S rRNA序列,采用稳定同位素探测技术(stable isotope probing,DNA-SIP)和显微放射自显影-荧光原位杂交技术(microautoradiographyfluorescence in situ hybridization,MAR-FISH)相结合,为分析尚不能培养的降解三氯生的微生物研究提供新思路[17]。Mota等[18]也指出16S rRNA荧光原位杂交在检测氨氧化和亚硝酸盐氧化微生物方面具有广泛的应用前景。以往的研究也发现,基于 16S rRNA序列的荧光原位杂交技术在鉴定海洋沉积物微生物,尤其是无法培养的微生物方面具有不可替代的优越性[19],且 ITS序列结合FISH在鉴定产毒真菌方面优势明显[20]。
2.2 16S rRNA/ITS和荧光定量PCR与微生物检测
荧光定量PCR是Applied Biosystems公司推出的通过荧光染料或探针标记的特异性探针对PCR产物进行跟踪的定量试验技术。荧光定量PCR技术具有实时监测的优越性,能够通过对产物及过程的分析计算模板的初始浓度。荧光定量PCR技术在微生物的鉴定、溯源及定量分析方面已有广泛的应用[21-22]。在采用荧光定量PCR分析16S rRNA单核苷酸多态性与结核分支杆菌耐药性的研究中发现 80.0%的结核分枝杆菌多耐药性可通过荧光定量PCR进行检测后分析获得[23]。郭毅等通过在白色念珠菌ITS2上设计Taq man探针建立了快速鉴定白色念珠菌的实时荧光定量PCR方法,且方法具有特异性和灵敏度高、重复性好的优点,适用于白色念珠菌的快速鉴定[24]。Libert等根据18S rDNA的内部转录间隔区1设计扩增引物,对环境样本的花斑曲霉进行有效分析[25]。荧光定量PCR在微生物的定量分析方面具有明显的优势,但其在鉴定微生物方面还存在一些地方需要改进、提高,如准确性方面[26]。
2.3 16S rRNA/ITS与HTS应用于微生物鉴定
16S rRNA/ITS结合HTS是目前微生物鉴定及分型的主要手段,且随着测序技术的成熟,其得到越来越广泛的应用[27]。在结合荧光定量PCR和DNA测序技术对引起败血症微生物鉴定的研究中证实二者结合对 16S rRNA基因序列分析比常规的实验室检测时间明显缩短,且能够比常规实验室检测提供更丰富的信息[28]。Lange等[29]在采用16S rRNA测序对葡萄球菌属进行种的鉴定时发现16S rRNA测序技术是一种客观、准确的葡萄球菌属鉴定方法,且研究人员通过该方法将分离的葡萄球菌分成13个种。Shin等采用16S rRNA全序列测序的方法对肠道微生物种群进行了分析,并指出16S rRNA全序列测序是一种快速、准确、有效的分析生物及临床样本中微生物种类的方法[30]。与上述报道类似,Kim等也通过对16S rRNA基因测序成功分析了放射治疗前后肠道菌群的变化[31]。但是16S rRNA基因测序技术也存在一些局限性:一些芽孢杆菌属的微生物如枯草芽孢杆菌、短小芽孢杆菌等存在16S rRNA序列的高度相似性,很难通过16S rRNA序列完成分型,而Mohkam等发现RAPD指纹图谱、rpoB及recA测序比16S rRNA序列分析具有更优越的优越性[32]。因此,16S rRNA序列分析应与其他方法结合进行微生物的鉴定分型。Soni等[33]通过16S rRNA结合hlyA基因测序鉴定了80例不同来源的单核细胞增生李斯特菌,包括英诺克李斯特氏菌、伊氏李斯特氏菌、西儿李斯特菌和威尔斯李斯特菌及格氏李斯特菌等。贾微等[34]也通过16S rRNA序列分析技术及RAPD指纹图谱分析对单核增生李斯特菌的母婴同源性进行了分析,证实母婴感染的单增李斯特菌株间属于同源。ITS序列分析结合高通量测序也为微生物鉴定研究提供新的手段,Ruegger等通过分析SSU与LSU rRNA基因之间间隔区序列,提供了一种改良的高通量测序分析 rRNA内部转录间隔区序列的研究细菌的方法[5]。
2.4 16S rRNA/ITS与基因芯片技术
基因芯片技术是通过与一组已知序列的核酸探针杂交后进行核酸序列测定的方法。目前,基因芯片技术已经与 16S rRNA序列分析相结合实现了多种微生物的鉴定[35-36]。16S-23S rRNA基因间隔的寡核苷酸序列具有种属的特异性,Kim等通过设计43个间隔序列特异性的寡核苷酸探针,准确鉴定了 708株与败血症相关的微生物,准确度达到 85.8%[36]。在采用基因芯片技术分析颅内感染患者脑脊液中病原菌种类分析时也发现基因芯片技术对颅内感染的诊断阳性率及脑脊液中金黄色葡萄球菌、肺炎克雷伯菌、大肠杆菌、肺炎双球菌的检出率高于常规细菌培养[37]。同时,董进浪等在16S rRNA基因保守区设计PCR反应的通用引物及革兰阳性菌、革兰阴性菌的通用探针,利用可变区的差异设计合成特异性探针,构建基因芯片均达到预期效果,具有简单、快速、特异性及敏感性高的特点[38]。
虽然 16S rRNA序列分析与分子生物学技术结合已实现了多种微生物的鉴定及分型,但其多集中于基础研究和临床应用,在药品微生物质量控制方面的报道较少。随着社会对食药质量关注度的不断提升和药品检验机构水平的进步,基于16S rRNA序列分析的分子生物学技术在药品质量控制领域经得到更广泛的应用。
3.1 水平基因转移(horizontal gene transfer,HGT)与多拷贝的异质性
HGT是在差异生物个体之间或单个细胞的细胞器之间进行的遗传物质的交流过程[39]。HGT打破了亲缘关系的界限,使得基因的流动更为复杂。早期的研究认为16S rRNA内不存在HGT,但近年来的研究发现,多种细菌 16S rRNA内发生了HGT的现象[40-42]。已有的证据显示,虽然核糖体rRNA的基因在三级结构上容易发生变异(变异率达 40.7%),甚至在蛋白结合位点也容易发生,但其二级结构是比较保守的,即其功能主要与其二级结构有关[43]。因此,在进行溯源分析及种群结构分析时应避免使用这些可发生 HGT的区域进行。在对噬藻体与其宿主蓝藻间的HGT以及遗传多样性进行研究时也发现噬藻体的光合作用基因最初来源于宿主蓝藻中,噬藻体能介导不同蓝藻之间的光合作用基因的交换,滇池中的蓝藻与噬藻体频繁的进行着光合作用基因的HGT[44]。
原核生物的 rRNA在基因组中通常是以多拷贝的形式存在,而同一菌种/株中不同拷贝的基因序列也可能存在差异性,即基因的异质性。在对1 690种细菌基因组进行分析时发现16S rRNA具有单拷贝的比例仅为 15%,而具有 3~7个拷贝的基因组为21%,16S rRNA中具有>1%差异的基因组达到2.4%[45]。
3.2 数据库完整性与某些种群的分辨率
16S rRNA/ITS序列分析进行微生物鉴定和分型通常基于已经发现并登陆的基因。由于GENEBANK为开放上传的数据库,其数据序列的准确性仍存在一定的疑问。对于16S rRNA基因序列的相似程度与细菌种属的相关性尚无统一定论。一些芽孢杆菌属的微生物如枯草芽孢杆菌、短小芽孢杆菌等存在 16S rRNA序列的高度相似性,很难通过16S rRNA序列完成分型[32]。不同种属间细菌可能发生基因水平的转移,并且与16S rRNA进行重组,形成嵌合体结构。因此,使用 16S rRNA探针或进行同源性序列分析时可能存在嵌合体结构。
药品无菌是药品对微生物限量的最高要求,是保障药品质量的重要环节。无菌检查的药品范围除了注射剂外,眼用制剂以及用于手术、严重烧伤和严重创伤的局部给药制剂也要求无菌。目前各国药典均要求不得对无菌制剂进行复试。因此,对样品中疑似检出菌进行溯源分析,判定其来源显得十分重要。分子生物学技术在药品微生物鉴定和分型方面已经有了广泛的应用和认可,而基于 16S rRNA基因序列分析是为药品微生物溯源分析的关键。郑小玲等[46]在生化鉴定仪的基础上采用16S rRNA基因测序鉴定了254株收集到的菌,初步建立了微生物检测实验室的环境菌库,为开展污染水平风险评估提供了参考。范一灵等也采用 16S rRNA基因测序结合生化分析仪、RiboPrinter系统和DiversiLab系统对14株葡萄球菌进行鉴定和溯源分析,为完善药品无菌检查过程控制和溯源调查提供解决方案[47]。已有的报道也指出单一的测序分析方法存在一定的缺陷,新一代宏基因组测序技术及分析方法需要进一步改进与完善,才能对环境微生物混合菌株进行快速、准确地鉴定,从而开展建库溯源工作[48]。对16S rDNA序列及其间隔区进行准确测序分析能够有效鉴别亲缘关系很近的微生物种,对微生物靶基因序列分析也有利于快速鉴定微生物[49]。在对复方益肝灵制剂中检出微生物进行分型溯源与产品质量评价时发现 16S rRNA测序和核糖体分型鉴定结果一致,且通过微生物鉴定分型能够为微生物监控、污染排查和追溯解决方案提供支持[50]。药品微生物限度检查时要求控制菌不得复验,而结合 16S rRNA测序和生化鉴定与血清学分型的结果一致,是控制菌检出鉴定的重要手段[51]。为鉴定乳酶生菌株并修订其质量标准,厉高慜等采用 16S rDNA全长序列测定不同来源乳酶生制剂中分离的菌株后发现,现行乳酶生质量标准中的肠链球菌应改名为屎肠球菌[52]。另外,选择高保守性的 16S rDNA区段序列分析能够对疫苗生产用种子进行质量控制,有效防止种子批的变异[53]。在对泛耐药鲍氏不动杆菌医院感染暴发的溯源调查发现该院NSICU发生的泛耐药鲍氏不动杆菌医院感染经16S-23S rRNA基因测序分析确认为医院感染暴发[54]。在对一起沙门菌引起的食源性疾病爆发的溯源分析中也发现,16S rRNA基因分型结果与PFGE分型方法的结果一致,实现了致病菌的准确溯源[55]。陈晨等[56]通过构建基于 16S rRNA序列的细菌分类鉴定在线分析平台,实现了快速、简单的对病原菌进行鉴定,有效提高传染病预防控制和感染性疾病临床诊疗中的病原菌鉴定和疫情溯源的速度。在对基于拟杆菌特异性16S rRNA基因的塘坝型饮用水污染溯源的研究中也发现,拟杆菌特异性16S rRNA基因的PCR-DGGE溯源方法结果可靠、操作简便,比大肠杆菌phoE基因的PCR-DGGE溯源方法更优[57]。裴晓燕等[58]利用基于 16S rRNA技术建立的数据库可以及时有效地比较不同地域克洛诺菌属分离株的相似性,并估计种群内部变异程度等,为克洛诺菌属食源性疾病的散发、暴发事件及常规监测中的追踪和溯源等提供有效的科学研究资料。笔者所在单位目前也进行了部分环境菌的收集和测序,初步建立了药品微生物检验实验室的环境菌库。
传统的经典微生物鉴定方法是微生物鉴定的不可或缺的手段,但其局限性亦越来越明显。随着高通量测序及分子生物学技术的发展,16S rRNA/ITS序列分析对微生物的鉴定、分析及溯源展现出不可比拟的优越性。但目前该方法也存在一定的局限性,如HGT及嵌和、数据库准确性等,会导致鉴定不准确或无法鉴定的现象,当出现16S rRNA序列分析无法鉴定或存在质疑时,需进行多种方法的交叉鉴定验证。但随着技术的进步和数据库的不断扩充,基于16S rRNA序列分析的分子生物学技术在药品质量控制领域的应用将日益广泛。
[1] RINKE C, SCHWIENTEK P, SCZYRBA A, et al. Insights into the phylogeny and coding potential of microbial dark matter [J]. Nature, 2013, 499(7459): 431-437.
[2] DENG S P, HUANG D S. An integrated strategy for functional analysis of microbial communities based on gene ontology and 16S rRNA gene [J]. Int J Data Min Bioinform,2015, 13(1): 63-74.
[3] XIA L P, BIAN L Y, XU M, et al. 16S rRNA gene sequencing is a non-culture method of defining the specific bacterial etiology of ventilator-associated pneumonia [J]. Int J Clin Exp Med, 2015, 8(10): 18560-18570.
[4] PEI A Y, OBERDORF W E, NOSSA C W, et al. Diversity of 16S rRNA genes within individual prokaryotic genomes [J].Appl Environ Microbiol, 2010, 76(12): 3886-3897.
[5] RUEGGER P M, CLARK R T, WEGER J R, et al. Improved resolution of bacteria by high throughput sequence analysis of the rRNA internal transcribed spacer [J]. J Microbiol Methods,2014(105): 82-87.
[6] SASHIDA H, SASAOKA F, SUZUKI J, et al. Two clusters among mycoplasma haemomuris strains, defined by the 16S-23S rRNA intergenic transcribed spacer sequences [J]. J Vet Med Sci, 2013, 75(5): 643-648.
[7] SULAIMAN I M, JACOBS E, SIMPSON S, et al. Molecular identification of isolated fungi from unopened containers of greek yogurt by DNA sequencing of internal transcribed spacer region [J]. Pathogens, 2014, 3(3): 499-509.
[8] CLEMENTINO M M, DE FILIPPIS I, NASCIMENTO C R,et al. Pcr analyses of trna intergenic spacer, 16S-23S internal transcribed spacer, and randomly amplified polymorphic DNA reveal inter- and intraspecific relationships of enterobacter cloacae strains [J]. J Clin Microbiol, 2001, 39(11): 3865-3870.
[9] LEBLOND-BOURGET N, PHILIPPE H, MANGIN I, et al.16S rRNA and 16S to 23S internal transcribed spacer sequence analyses reveal inter- and intraspecific bifidobacterium phylogeny [J]. Int J Syst Bacteriol, 1996, 46(1): 102-111.
[10] CIARDO D E, LUCKE K, IMHOF A, et al. Systematic internal transcribed spacer sequence analysis for identification of clinical mold isolates in diagnostic mycology: A 5-year study [J]. J Clin Microbiol, 2010, 48(8): 2809-2813.
[11] TANASUPAWAT S, KOMMANEE J, YUKPHAN P, et al.Identification of acetobacter strains from thai fermented rice products based on the 16S rRNA gene sequence and 16S-23S rRNA gene internal transcribed spacer restriction analyses [J].J Sci Food Agric, 2011, 91(14): 2652-2659.
[12] THIERY O, VASAR M, JAIRUS T, et al. Sequence variation in nuclear ribosomal small subunit, internal transcribed spacer and large subunit regions of rhizophagus irregularis and gigaspora margarita is high and isolate-dependent [J]. Mol Ecol, 2016, 25(12): 2816-2832.
[13] BOSSHARD P P, ABELS S, ZBINDEN R, et al. Ribosomal rna sequencing for identification of aerobic gram-positive rods in the clinical laboratory (an 18-month evaluation) [J]. J Clin Microbiol, 2003, 41(9): 4134-4140.
[14] ZHANG G L, JING R X. The application of microorganism identification and genotyping techniques in microbial contamination detection in food and drug quality control [J].Chin J Mod Appl Pharm(中国现代应用药学), 2014, 31(11):1412-1417.
[15] PETRICH A, ROJAS P, SCHULZE J, et al. Fluorescence in situ hybridization for the identification of treponema pallidum in tissue sections [J]. Int J Med Microbiol, 2015, 305(7):709-718.
[16] KARLSSON F, KLITGAARD K, JENSEN T K. Identification of treponema pedis as the predominant treponema species in porcine skin ulcers by fluorescence in situ hybridization and high-throughput sequencing [J]. Vet Microbiol, 2014, 171(1/2):122-131.
[17] LOLAS I B, CHEN X, BESTER K, et al. Identification of triclosan-degrading bacteria using stable isotope probing,fluorescence in situ hybridization and microautoradiography[J]. Microbiology, 2012, 158(Pt 11): 2796-2804.
[18] MOTA C R, SO M J, DE LOS REYES FL. Identification of nitrite-reducing bacteria using sequential mRNA fluorescence in situ hybridization and fluorescence-assisted cell sorting [J].Microb Ecol, 2012, 64(1): 256-267.
[19] DEKAS A E, ORPHAN V J. Identification of diazotrophic microorganisms in marine sediment via fluorescence in situ hybridization coupled to nanoscale secondary ion mass spectrometry (fish-nanosims) [J]. Methods Enzymol,2011(486): 281-305.
[20] LEZAR S, BARROS E. Oligonucleotide microarray for the identification of potential mycotoxigenic fungi [J]. BMC Microbiol, 2010(10): 87. doi: 10.1186/1471-2180-10-87.
[21] PARRA E, SEGURA F, TIJERO J, et al. Development of a real-time PCR for bartonella spp. Detection, a current emerging microorganism [J]. Mol Cell Probes, 2017(32):55-59.
[22] ZRIMEC J, KOPINC R, RIJAVEC T, et al. Band smearing of pcr amplified bacterial 16S rRNA genes: Dependence on initial pcr target diversity [J]. J Microbiol Methods, 2013,95(2): 186-194.
[23] BLASCHITZ M, HASANACEVIC D, HUFNAGL P, et al.Real-time pcr for single-nucleotide polymorphism detection in the 16S rRNA gene as an indicator for extensive drug resistance in mycobacterium tuberculosis [J]. J Antimicrob Chemother, 2011, 66(6): 1243-1246.
[24] GUO Y, YANG J X, SHAO D H, et al. A novel Real-time PCR assay based on the internal transcribed spacer 2 for rapid identification of Candida Albicans [J]. J Med Res(医学研究杂志), 2016, 45(7): 79-84.
[25] LIBERT X, CHASSEUR C, BLADT S, et al. Development and performance assessment of a qualitative sybr(r) green real-time pcr assay for the detection of aspergillus versicolor in indoor air [J]. Appl Microbiol Biotechnol, 2015, 99(17):7267-7282.
[26] CALLBECK C M, SHERRY A, HUBERT C R, et al.Improving pcr efficiency for accurate quantification of 16S rRNA genes [J]. J Microbiol Methods, 2013, 93(2): 148-152.
[27] LI D P, GUO M Z, XU W T. Advances and applications on methodology of 16S rRNA sequencing in Gut Microbiota analysis [J]. Biotechnol Bull(生物技术通报), 2015, 31(2):71-77.
[28] MIDAN D A, ABO EL FOTOH W M, EL SHALAKANY A H. The potential role of incorporating real-time pcr and DNA sequencing for amplification and detection of 16S rRNA gene signatures in neonatal sepsis [J]. J Matern Fetal Neonatal Med,2017, 30(12): 1476-1483.
[29] LANGE C C, BRITO M A, REIS D R, et al. Species-level identification of staphylococci isolated from bovine mastitis in brazil using partial 16S rRNA sequencing [J]. Vet Microbiol,2015, 176(3/4): 382-388.
[30] SHIN J, LEE S, GO M J, et al. Analysis of the mouse gut microbiome using full-length 16S rRNA amplicon sequencing[J]. Sci Rep, 2016(6): 29681. Doi: 10.1038/srep29681.
[31] KIM YS, KIM J, PARK S J. High-throughput 16S rRNA gene sequencing reveals alterations of mouse intestinal microbiota after radiotherapy [J]. Anaerobe, 2015(33): 1-7.
[32] MOHKAM M, NEZAFAT N, BERENJIAN A, et al.Identification of bacillus probiotics isolated from soil rhizosphere using 16S rRNA, reca, rpob gene sequencing and rapd-pcr [J]. Probiotics Antimicrob Proteins, 2016, 8(1): 8-18.
[33] SONI D K, DUBEY S K. Phylogenetic analysis of the listeria monocytogenes based on sequencing of 16S rRNA and hlya genes [J]. Mol Biol Rep, 2014, 41(12): 8219-8229.
[34] JIA W, SUN M Y, AN F X, et al. Study of 16S rRNA detection and RAPD fingerprinting to identify Listeria monocytogenes causing maternal and neonatal infections [J]. J Path Bio(中国病原生物学杂志), 2015, 10(6): 503-507.
[35] DONG J L, LI H, WANG J, et al. Construction of gene chip and its application in bacteria pneumonia [J]. Chin J Lab Diagn(中国实验诊断学), 2014, 18(7): 1048-1052.
[36] KIM C M, SONG E S, JANG H J, et al. Development and evaluation of oligonucleotide chip based on the 16S-23S rRNA gene spacer region for detection of pathogenic microorganisms associated with sepsis [J]. J Clin Microbiol, 2010, 48(5):1578-1583.
[37] 丁锦荣, 管义祥, 吴德模, 等. 56例疑诊颅内感染患者脑脊液中病原菌的基因芯片技术检测结果[J]. 山东医药, 2014,54(35): 83-85.
[38] DONG J L, LI H, WANG J, et al. Construction of gene chip and its application in bacteria pneumonia [J]. Chin J Lab Diagn(中国实验诊断学), 2014, 18(7): 1048-1052.
[39] LIU C, LI J B, RUI J P, et al. The application of the 16S rRNA gene in microbial ecology: current situation and problems [J].Acta Ecologia Sin(生态学报), 2015, 35(9): 2769-2788.
[40] KINGSTON A W, ROUSSEL-ROSSIN C, DUPONT C, et al.Correction: Novel reca-independent horizontal gene transfer in escherichia coli k-12 [J]. PLoS One, 2015, 10(11): e0143991.
[41] MILLER J H, NOVAK J T, KNOCKE W R, et al. Survival of antibiotic resistant bacteria and horizontal gene transfer control antibiotic resistance gene content in anaerobic digesters [J].Front Microbiol, 2016(7): 263. doi: 10.3389/fmicb.2016.00263
[42] TIAN R M, CAI L, ZHANG W P, et al. Rare events of intragenus and intraspecies horizontal transfer of the 16S rRNA gene [J]. Genome Biol Evol, 2015, 7(8): 2310-2320.
[43] KITAHARA K, YASUTAKE Y, MIYAZAKI K. Mutational robustness of 16S ribosomal RNA, shown by experimental horizontal gene transfer in escherichia coli [J]. Proc Natl Acad Sci USA, 2012, 109(47): 19220-19225.
[44] LIU T T. The horizontal gene transfer between cyanophage and cyanobacteria and their genetic diversity [D]. Kunming:Kunming University of Science and Technology, 2012.
[45] VETROVSKY T, BALDRIAN P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses [J]. PLoS One, 2013, 8(2):e57923.
[46] ZHANG X L, WANG Z N, WANG Z J, et al. Establishment of the environmental microbial library in the drug sterility testing laboratory [J]. Chin J Mod Appl Pharm(中国现代应用药学),2015, 32(7): 847-850.
[47] FAN Y L, JIANG B, FANG R, et al. Identification and characterization of bacterialcontaminations isolated from drug sterility test [J]. Chin J Pharm Anal(药物分析杂志), 2011,31(6): 1067-1072.
[48] ZHENG X L, WANG Z J, LI Y, et al. Application of a variety of sequencing technology in the identification of environmental microorganisms [J]. Chin J Pharm Anal(药物分析杂志), 2016, 36(1): 46-52.
[49] HAKOVIRTA J R, PREZIOSO S, HODGE D, et al.Identification and analysis of informative single nucleotide polymorphisms in 16S rRNA gene sequences of the bacillus cereus group [J]. J Clin Microbiol, 2016, 54(11): 2749-2756.
[50] FENG Z, ZHONG W, LIU D L, et al. Microorganism identification and molecular ribotyping in quality control of Yiganling oral tablets [J]. Chin J Anal(药物分析杂志), 2015,35(12): 2083-2088.
[51] ZHENG X L, YANG D Y, LI Y, et al. Comparison of modern strain identifation technologies in drug control bacteria test [J].Chin J Health Lab Tec(中国卫生检验杂志), 2015, 25(5):658-660.
[52] LI G M, ZHAO L, NIE L, et al. Identification of lactasin strains by full length 16S rDNA sequence analysis and biochemical tests [J]. Chin J Anal(药物分析杂志), 2014, 34(5):775-781.
[53] YAO S S, WANG W, JI G X, et al. 16S rRNA gene sequencing of four domestic Salmonella paratyphiA vaccine strains [J]. Chin J Biologicals(中国生物制品学杂志), 2008,21(7): 595-597.
[54] GUO Y Q, ZHANG J, ZHONG X Z, et al. Tracing the source of an outbreak of pan-drug resistant Acinetobacter baumannii[J]. Chin J Nosocomiol(中华医院感染学杂志), 2016, 26(20):4579-4582.
[55] SHI W Y, ZHENG Y K, YE Z Y, et al. Souce tracing of one outbreak of foodborne disease caused by Salmonella [J]. Prog Mod Biomed(现代生物医学进展), 2015, 15(31): 6043-6045.
[56] CHEN C, PENG K, WANG H Y, et al. An online platform SSUDB: database of bactiria identification and classification with 16S rDNA [J]. Dis Surveil(疾病监测), 2013, 28(3):236-240.
[57] ZHANG X, ZHU C X, FENG G D, et al. Potential use of bacteroidales specific 16S rRNA in tracking the rural pond-drinking water pollution [J]. J Agro-Environ Sci(农业环境科学学报), 2011, 30(9): 1880-1887.
[58] PEI X Y, GUO Y C, ZHOU Z, et al. Establishment of a source tracing database for Cronobacter spp. (Former Enterobacter sakazakii) [J]. Chin J Food Hygiene(中国食品卫生杂志),2010, 22(2): 105-108.
Application of 16S rRNA/ITS Sequencing and Analysis in the Identification, Classification of Microbial and Drug Quality Control
ZHANG Guolin1,2, SU Yuanke3, SHEN Haiying1, GU Min1, XING Yiwen1
(1.Suzhou Institute for Drug Control,Suzhou 215000, China; 2.Nanjing University of Traditional Chinese Medicine, Nanjing 210000, China; 3.Longkou Entry-Exit Inspection and Quarantine Bureau, Longkou 265700, China)
ABSTRACT:Traditional methods for identification and classification of microorganisms have limitations in those of unculturable, slow-growth and special dietary needed bacteria. Fully automatic bacteria identification and drug susceptibility analysis system and MALDI-TOF MS are belonged to phenotype identification which is dependent obviously on the database of equipment. These two methods are defective in the variety kinds of environmental microorganisms. Modern molecular based identification methods supply new idea for the environmental microorganism analysis. 16S rRNA/ITS, a sequence contains conservative and variant region is not related with the culture state is widely used in the identification and classification of microorganisms. Microorganism is a significant factor for drug quality control. The present dissertation tried to explore the structure of 16S rRNA/ITS and its application on drug quality control area.
KEY WORDS:microorganism identification; trace-back investigation; morphological identification; 16 Small Subunit Ribosomal RNA; internal transcribed spacer; molecular biological technique
REFERENCES
中图分类号:R915
文献标志码:B
文章编号:1007-7693(2017)10-1489-07
DOI:10.13748/j.cnki.issn1007-7693.2017.10.030
引用本文:张国林, 苏远科, 沈海英, 等. 16S rRNA/ITS基因序列分析与微生物分类鉴定及其在药品微生物质量控制中的应用[J]. 中国现代应用药学, 2017, 34(10): 1489-1495.
基金项目:苏州市科技发展计划项目(SYS201425);山东省出入境检验检疫局项目(SK201618)
作者简介:张国林,男,博士,副主任药师 Tel: (0512)67079902 E-mail: zhangguolin2006@163.com 共同第一作者:苏远科,男,硕士,工程师 Tel: (0512)67079902 E-mail: 414589831@qq.com
收稿日期:2017-01-12
(本文责编:蔡珊珊)